
Abstract
Nowadays, risperidone is an atypical antipsychotic drug that has been increasingly used for treatment and maintenance therapy in schizophrenia. However, partially affected by genetic or environmental factors, there is significant difference in treatment outcomes among patients. In this study, we aimed to interpret the difference between good and poor responders treated with risperidone in both genetic and epigenetic levels in 288 mainland Chinese patients. We recruited a Henan cohort including 98 patients as initial discovery group and then confirmed our results in Shanghai cohort. In genetic studies, we found 10 candidate single-nucleotide polymorphisms (SNPs) and 2 rare variants in Henan cohort by next-generation sequencing of 100 risperidone-response-related genes. After replication in Shanghai cohort by massarray platform, ultimately, rs6706232 and rs4818 were significantly associated with risperidone response in the two cohort meta-analysis (P=0.024 and 0.04, respectively). Besides, we also selected another reported 17 candidate SNPs associated with risperidone drug response to replicate in our mainland Chinese samples, while, we found no significant SNPs after Bonferroni correction. In epigenetic studies, we investigated the methylation status in promoters or gene-coding region of risperidone drug response-related genes including CYP3A4, CYP2D6, ABCB1, HTR2A, DRD2. Totally we found seven significant CpG sites in the meta-analysis with Bonferroni-corrected PCYP3A4_CpG_-36=0.0014, PCYP3A4_CpG_-258=0.0013, PCYP3A4_CpG_-296=0.0014, PCYP3A4_CpG_-367:-372:-374=0.028, PCYP2D6_CpG_193=0.012, PCYP2D6_CpG_242:244:250=0.00076 and PCYP2D6_CpG_284=0.034, respectively. As genetic and epigenetic factors may interactively affect drug response, we finally carried out a multivariant interaction analysis with multifactor dimensionality reduction and discovered a significant four-locus model (CYP3A4_CpG_-82:-86 +rs6280+rs1800497+rs6265, P=0.038) affecting drug response. These findings could partially explain different risperidone response outcome in Chinese population in a systematic level.
Similar content being viewed by others
Introduction
Risperidone is an antipsychotic drug that has been increasingly used for treatment and maintenance therapy in schizophrenia and related psychotic disorders.1 Risperidone is metabolized to its active metabolite, 9-hydroxyrisperidone (9-OH-risperidone with potent antagonistic properties for the dopamine D2 and serotonin 5-hydroxytryptamine-2 (5-HT2) receptors). However, there are significant interindividual differences in clinical response and side effects, meanwhile, optimizing drug treatment for patient is often by trial and error which costs a lot of time and money, so it is crucial to identify more novel drug-response-related markers to predict drug response.
The variability in the risperidone response can be caused by genetic, epigenetic, physiologic and environmental factors. Genetic factors are mostly assumed to have a close relationship with drug treatment response,2 and on this basis, a number of pharmacokinetic studies have been performed. To date, most studies have typically focused on candidate genes, mainly selected drug metabolizing enzyme genes, transport genes, neurotransmitter receptors genes, such as dopamine or serotonin receptors. Several studies have shown positive associations between genetic variation and risperidone response, for example, CYP2D6, ABCB1, dopamine receptors and serotonin receptors have ever been reported to be significant associated with risperidone’s efficiency and risperidone-induced adverse effects.3, 4, 5 However, these studies mainly focused on a few genes through candidate gene association study method and most were performed in small sample size without independent replication, because of which these results could not been confirmed by different groups and used in clinical practices. In recent years, 14 candidate genes have been identified in relation to risperidone treatment response in a genome-wide association study.6 Although this study was also performed in relatively small sample size, it has the low power to uncover the typical genome-wide association study significant variants and sometimes the genome-wide association study results are not replicated across other studies or populations.
In addition, epigenetic factors can also affect drug treatment by modulating the expression of key genes involved in the metabolism and distribution of drugs as well as drug targets, thereby contributing to interindividual variation in drug response,7 which mainly derived from DNA methylation modification changes. There have been some reports that DNA methylation status may serve as a pharmacogenomics biomarker. More recently, DNA methylation in drug absorption, distribution, metabolism, excretion (ADME) genes such as GSTP1, GPx3, ABCB1, ABCG2 and the nuclear receptor vitamin D receptor have been reported to be associated with drug response,8, 9, 10, 11 which reflects a strong potential of epigenetic marks to serve as predictors of antipsychotic drug response. However, epigenetic studies of risperidone response have rarely been reported. Furthermore, the drug response of risperidone involves complex drug ADME-related molecular networks and pathways; as a result, the common variants associated with risperidone may have smaller effect sizes, and they may predict a response only when combined variants in genes in a known molecular pathway test whether the pathway is associated with the phenotype. This pathway-based association approach provides a more powerful strategy for pharmacogenomics study.
In this study, to comprehensively discover the predictor of risperidone response, we conducted the pharmacogenomics study using target sequencing technology and epigenetic study using massARRAY technology. Also, we performed combined analysis of different markers and replication studies in independent subjects, which laid the foundation for comprehensively discovering the predictor of risperidone response in Chinese population.
Materials and methods
Subjects
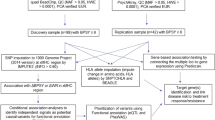
Two cohorts of Chinese Han in-patients with schizophrenia were enrolled in the present study. Figure 1 is research workflow. One hundred and ninety patients were recruited from Shanghai Mental Health Center, 98 patients were recruited from Henan Provincial Mental Health Center. All patients recruited met the following criteria: (1) they satisfied the Diagnostic and Statistical Manual of Mental Disorders-IV (DSM-IV) criteria for schizophrenia; (2) they had no physical complication or other substance abuse; (3) they had no history suggesting resistance to antipsychotic drug treatment; (4) they had not received any medication for 4 weeks; and (5) they had not previously received second-generation antipsychotics. The study protocol was drawn up according to the principles of the Helsinki Accord and was reviewed and approved by the Shanghai Ethical Committee of Human Genetic Resources. The statement of informed consent was obtained from all the subjects after full explanation of the procedure. Genomic DNA was extracted by QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) and quantified on three platforms including NanoDrop 2000 (Thermo, Wilmington, DE, USA), Qubit Fluorimeter (Invitrogen, Eugene, OR, USA), 2100 Bioanalyzer (Agilent, Waldbronn, Germany) according to manufacturer’s protocol.

Research workflow of this study. ADME, absorption, distribution, metabolism, excretion; MDR, multifactor dimensionality reduction; NGS, next-generation sequencing; SNP, single-nucleotide polymorphism.
Clinical assessment
For the recruited subjects, the dosage of risperidone was 2 mg per day initially and then gradually increased to 4 mg per day within the first week, which was maintained until the end of week 2. After that, the dosage was adjusted according to individual tolerance. For all the participants, medication compliance was closely monitored and confirmed by nursing staff, and no other medication was given except bedridden for extrapyramidal side effects, flunitrazepam for insomnia and sennoside for constipation during the study period.
Clinical effect was assessed on the Positive and Negative Syndrome Scale (PANSS), including the positive, negative and general psychopathology subscales. For the recruited patients, clinical assessments were conducted on the day of admission, as well as at the end of week 4. In each cohort, all PANSS ratings were conducted independently by two qualified psychiatrists, who were blind to the genotype of patients. And the inter-rater reliability between the two psychiatrists is good. Risperidone treatment efficacy was measured in terms of the reduction in PANSS scores. For the independent samples, risperidone response was classed into four groups, cured (⩾75%), significant progress (50–74%), progress (25~49%) and ineffective (<25%) based on PANSS scores.
Methylation analysis
We screened CpG-rich spot in the upstream of promoter region or within candidate genes including CYP3A4, CYP2D6, HTR2A, ABCB1 and DRD2 by UCSC Human Genome Browser Gateway (http://genome.ucsc.edu/cgi-bin/hgGateway), and designed specific PCR primers for bisulfate treatment amplification by EpiDesigner software (www.epidesigner.com). We detected CpG methylation status on MassARRAY Analyzer 4 platform and analyzed using software Epityper1.0.5 (Sequenom, San Diego, CA, USA).
Next-generation sequencing
The systematic association study between exon polymorphisms of candidate genes and response to risperidone in Chinese Han schizophrenia patients was carried out with Miseq pair-end sequencing technology after HaloPlex Target Enrichment system (HaloPlex Custom Kits, 1–500 kb, ILMFST, 96, G9901B). The primers were designed on SureDesign software (http://earray.chem.agilent.com/suredesign/, Agilent) and data analysis was performed on SureCall (www.genomics.agilent.com) software. The result of next-generation sequencing (NGS) was validated by iPLEX Gold SNP method on MassARRAY Analyzer 4 platform.
Statistical analysis
Statistical analysis was carried out as described previously12 with minor modifications. Cohort characteristics including gender, age and weight differences of demographic and clinical variables were examined first to affirm the homogeneity of the good responders and poor responders in our samples with Student’s t-test. The reduction of the total and subscale scores of PANSS was used as a measure of clinical improvement of risperidone treatment. SPSS for Windows, version 11.0 (SPSS, Chicago, IL, USA) was used for statistical analysis.
To substantiate the results, allele and genotype frequencies of each polymorphism were compared between good-responders and poor-responders groups using the χ2 test on the online software SHEsis (http://analysis.bio-x.cn).13 And clinical good responders were defined as patients with 50% or even higher reduction in PANSS scores than the average level of all subjects; correspondingly, poor responsers were defined as patients with lower reduction than 50% in PANSS scores. All tests were two-tailed and statistical significance was assumed at P<0.05.
The mutual interactions between methylation sites and single-nucleotide polymorphisms (SNPs) were analyzed on multifactor dimensionality reduction (MDR) software as previously described.14 In the configuration file, 10-fold cross-validation was defined and the threshold ratio was set at 1.0. We ran the analysis 10 times using constant random number seeds and the results were averaged to avoid spurious outcomes due to chance divisions of the data. This MDR procedure can be carried out for each possible model size, if computationally feasible. Due to computation restrictions, we considered two-locus interactions through four-locus interactions. We determined statistical significance by comparing the average prediction error from the observed data with the distribution of average prediction errors under the null hypothesis of no association derived empirically from 1000 permutations. The null hypothesis was rejected when the upper-tail Monte Carlo P-value derived from the permutation test was <0.05.
Results
Patients characteristics
Detailed information about patients and research workflow could be found in Table 1 and Figure 1, respectively. There is no gender, age and weight difference between risperidone good-response group and poor-response group either in the Henan cohort or in the Shanghai cohort or in the whole sample. A power calculation indicated that we had the power of 80.9% to detect many of the SNPs with effect size =0.167 and d.f. =1 in the combined cohort of Henan and Shanghai.
The discovery and validation of genetic variant biomarkers associated with risperidone response
A hundred candidate genes that related with risperidone response were introduced in our study (Supplementary Table 1). In target sequencing, average read depth was 63.7 ×, and 1 ×, 8 ×, 20 × coverage with Q30 were 94.35%, 87.70%, 74.34%, respectively. A total of 330 SNPs were found by plink software with filtration conditions such as MAF (minor allele frequency) >0.01, Hardy–Weinberg equilibrium P>0.001, call rate >95% and then 10 SNPs were significantly associated with risperidone treatment response by target sequencing after data quality control and tested with generalized linear regression model with non-genetic confounding factors (age, onset age, weight) as covariants (Table 2). However, after P-value correction, all the 10 SNPs were no longer significant statistically (P>0.05). Second, to validate the technical accuracy of Miseq sequencing platform, we performed MassARRAY analysis in 80 Henan samples with specific primers (Supplementary Table 2) and the average accordance of the two platforms reached 94.88%, which guaranteed a high credibility of our data (data not shown). Besides, when we neglected the non-genetic confounding factors (age, onset age, weight), we found two mutations with low frequency (1%< MAF <5%), which were significantly different in risperidone treatment response (Supplementary Table 3). Third, in order to validate whether the 10 candidate SNPs detected by NGS remain significant, based on MassARRAY platform, we first tested the results in a smaller Henan cohort and then we found that five SNPs and four genotypes were significantly associated with risperidone treatment response without P-value correction; then we used a larger Shanghai cohort for further validation. A meta-analysis was carried out in the whole cohort, two SNPs and one genotype showed statistical significance before P-value correction ultimately (Table 3). For comparison, we included another 17 reported risperidone-response-related SNPs for validation in our cohort, while we only validated two significant SNPs in Henan cohort and two genotypes in meta-analysis without P-value correction (Table 4).
The discovery and validation of DNA methylation biomarkers associated with risperidone response
The PCR primers for bisulfate-treated amplification were listed (Supplementary Table 4). Significant CpG sites within CYP3A4 gene promoter region or CYP2D6 gene body were found associated with risperidone treatment (Figures 2, 3, 4), whereas no significant CpG sites in HTR2A, ABCB1, DRD2 gene promoters, respectively (data not shown).

Methylation rate of CYP3A4 and CYP2D6 gene promoter and gene-coding region in Henan Cohort. P*: P-value after Bonferroni correction; CYP3A4_CpG_-36: 36 bases ahead of CYP3A4 transcriptional start site; CYP2D6_CpG_36: 36 bases after CYP2D6 transcriptional start site.

Methylation rate of CYP3A4 and CYP2D6 gene promoter and gene-coding region in Shanghai Cohort as a replication group for validation of the results found in Henan cohort.

Meta-analysis about the methylation rate of CYP3A4 and CYP2D6 gene promoter and gene-coding region in Henan and Shanghai Cohort. Ten CpG sites were significantly related with risperidone treatment response; whereas after Bonferroni correction, there remained seven significant CpG sites between good responders and poor responders.
Multivariate interactions analysis of genetic variant and DNA methylation biomarkers associated with risperidone response
MDR software was used for detecting factor–factor interactions (including all SNPs, CpG rates, sex, age, weight and so on) in genetic case–control studies as having great advantages vs the conventional statistical approaches. We combined all the SNPs and CpG sites into MDR and built two-, three- and four-level interaction models with MDR software, then we found a best four-locus model (CYP3A4_CpG_-82:-86+rs6280+rs1800497+rs6265)with a testing-balanced accuracy of 58.94% and a cross-validation consistency of 7/10. The permutation testing showed that the four-locus model was significant with P=0.044 (Table 5).
Discussion
To provide the most effective and safe treatment for each schizophrenic patient, understanding the internal and external factors affecting response to antipsychotic drugs is important. Most studies put emphasis on either genetic or environmental aspect alone instead of combining both pharmacogenetics and pharmacoepigenetics. This prompted us to perform a more systematic study using two methods: next-generation sequence of 100 risperidone treatment response-related genes and epigenetic research five representative of these genes, including drug metabolic enzyme genes CYP450s (phase I), Uridine 5′-diphosphate-glucuronosyltransferases (UGTs; phase II), mostly studied receptor genes, other potentially related genes and five risperidone tightly associated ADME gene promoters or coding regions, which might optimize clinical treatment in antipsychotic drugs treatment. More importantly, most of the reported findings remain inconclusive and require either clinical examination or a larger clinical sample to validate, so we selected 17 other SNPs from literatures for validation and comparison of our NGS results based on Chinese Han population background.
NGS study
In NGS study, we found 10 candidate SNPs significantly related with risperidone treatment response in six genes (Table 2). We also confirmed NGS results by MassARRAY genotyping platform with an accuracy of 94.88% accordance in average (data not shown), whereas the significance of 10 candidate SNPs discovered by NGS platform no longer demonstrated significance after Bonferroni correction. As Henan cohort only contained less than 100 cases, we recruited another Shanghai cohort for the validation of NGS results and implemented a meta-analysis by combining the two cohorts (Table 3). Ultimately, we discovered two rare variant SNPs with 1%< MAF <5%, though both of them had limited impact in downstream protein functions predicted by protein online databases. We could not ignore the high effect score predicted by PolyPhen2; in addition, we found two SNPs, rs6706232 in UGT1A3 and rs4818 in catechol-O-methyltransferase (COMT) were significantly associated with risperidone treatment in meta-analysis (P=0.024 and P=0.04, respectively).
UGT1A3
UGTs are phase II drug metabolism enzymes in human tissues, containing nine functional isoforms including (UGT1A1, UGT1A3–UGT1A10) and four pseudogenes (UGT1A2, UGT1A11–UGT1A13). Among these enzymes, UGT1A3 has a critical role in endobiotic and xenobiotic compounds metabolism by catalyzing the glucuronidation of endogenous compounds such as bilirubin, bile acids, thyroid hormone, steroid hormones and substantial exogenous substrates such as many therapeutic drugs, heterocyclic and polycyclic hydrocarbons, and heterocyclic amines,15, 16, 17 Polymorphisms in UGT1A3 genes (http://www.pharmacogenomics.pha.ulaval.ca/cms/ugt_alleles/) could have significant influence on metabolism of endogenous compounds like bilirubin or variability in response to irinotecan among other drugs.18, 19 For example, effect of rs6706232 on OTS167 (a novel synthetic anticancer agent molecule undergoing clinical development) glucuronidation formation rates was merely modest suggesting this SNP may not significantly contribute to OTS167 clearance.20 In our study, rs6706232 was first found related with risperidone treatment in mainland Chinese Han population, allele A accounted for a higher proportion in good responders compared with poor responders at 33.3%, 24.6%, 25.2% in the Henan, Shanghai and total cohort, respectively, which means the G>A mutation might have certain impact on risperidone therapy. Recent research found that rs6706232 was significantly associated with UGT1A3 gene transcription and activity.21 Whether enzyme UGT1A3 directly, or its derivatives, affects risperidone response remains elusive although aromatic heteropolycyclic risperidone was cataloged a potential target catalyzed by UGT1A3 enzyme.
COMT polymorphisms
COMT, first discovered in 1957,22 has always been regarded as susceptibility gene in schizophrenia for its function of degrading catecholamine such as dopamine, epinephrine and norepinephrine.23, 24 Polymorphisms in COMT affected enzyme activity, for example, well-studied variant rs4680 G>A (substitution of valine to methionine at codon 158) resulted in decreased enzyme activity, which contributed to inefficient catalyzation and accumulation of dopamine, thus inducing positive symptoms in schizophrenia or even drug treatment failure.25, 26 Gupta et al.27 found that haplotype rs4818 and rs4680 in exon 4 was related with risperidone treatment response (P=0.028) in 398 schizophrenia patients and 241 healthy individuals from a homogeneous South Indian population. Recently, our group found that rs4818 was significantly associated with quetiapine drug response (P=0.00081) in the Shanghai cohort, but there was no correlation with risperidone drug response (P=0.515),28 whereas in this study, we confirmed the significance of rs4818 in 288 risperidone-treated schizophrenia patients (P=0.04) with similar G/C allele frequency in good responders or poor responders. The difference might partially result from different sample size or drug metabolism pathways and indications. Risperidone has a higher pituitary D2 receptor occupancy, which is suitable for positive symptoms than quetiapine, whereas quetiapine has various affinities for cerebral serotonergic(5-HT), D2 dopaminergic, histaminergic(H1) and adrenergic receptors, including much wilder indications than risperidone that involved more genetic pathways.29, 30 As a synonymous variant, rs4818 may have a milder influence on COMT enzyme function compared with rs4860. Another interesting result is that most significant polymorphisms were simultaneously found in COMT including rs4633, rs4680 and rs4818 (based on NGS), rs9606186 (based on MassARRAY) with P=0.018, 0.028, 0.039, 0.041 in the Henan cohort. The discovery of rs9606186, rs4680 and rs4818 also confirmed previous findings in Chinese patients carried out by our group.31 Taken together, these results suggest that COMT polymorphisms contribute greatly to risperidone treatment.
Replication of 17 SNPs
Replication of other reported 17 SNPs was carried out preliminarily in the Henan cohort and we found three candidate SNPs including rs4483927 in HRH4, rs6295 in 5HTR1A, rs9606186 in COMT with P=0.005, 0.009, 0.026, respectively, while rejected by multiple testing corrections. However, after validation in the Shanghai cohort for replication and meta-analysis, none of the three SNPs remained significant. This might partially attribute to different population background such as geography, nutrition32, 33 and so on, and the fact of this study using a small sample size also accounts for the inconsistent results in our study and others. This promotes us to further expand our sample size to nearly 600 qualified subjects for discovering the effect markers.
Comparison of previous results by our group
We compared this study with other pharmacogenomics results found by our group in the last decade to investigate the accordance of different studies, which might make complement or new clues to pharmacogenomics research. Previously, our group found no significant SNPs in CYP2D6, CYP3A4, CYP2E1 associated with risperidone treatment response,34, 35, 36 however, SNPs in CYP2D6, CYP3A4 and CYP2E1 in this study were not overlapped and rs2515641 in CYP2E1 was found significantly related with risperidone treatment response in the Henan cohort using the NGS method (P=0.007). Rs2515641, located in the eighth exon of CYP2E1 as a synonymous mutation (Phe421Phe), was first uncovered in the scanning of CYP2E1 in Chinese mainland Han population in 2010 (ref. 37; MAF=15.1%) and might relate to acute rejection in kidney transplantation recipients.38 However, the significance was not observed in the Henan and Shanghai cohort or meta-analysis when conducted in MassARRAY platform (P>0.05, data not shown). Besides, Xing, et al.39 found that the wild-type TT of rs1128503 in ABCB1 (synonymous mutation) carriers had better risperidone treatment response than other genotype carriers in 130 Chinese schizophrenia patients (F=3.976, P=0.021). But we failed to validate this SNP in meta-analysis, which might reflect the limited role in risperidone treatment response though the brain entry of risperidone and 9-OH-risperidone is greatly limited by ABCB1 product: p-glycoprotein. Meanwhile, loci rs6280 (Ser9Gly) of DRD3 gene has shown significant association with risperidone treatment response in the Henan cohort by the NGS method (adjusted P=0.031) or MassARRAY platform (adjusted P=0.015), and more allele C carriers in poor responders group than good responders with 41.8% vs 24.2% (Henan cohort, NGS platform), 52% vs 32% (Henan cohort, MassARRAY), which might be related with therapy response, but this result was inconsistent with Xuan et al.40
Epigenetic study
The evidence that epigenetic events can have an important role in regulating the expression of drug ADME genes, drug transporters, nuclear receptors and drug targets strongly implied that interindividual differences in their epigenetic status can contribute to the clinically observed variability in drug response,41 which could not be explained by genetic polymorphisms alone. We chose gene promoter or gene body region42 of five genes (CYP3A4, CYP2D6, HTR2A, DRD2, ABCB1) to investigate the correlation between their methylation status and risperidone treatment response in the Henan and Shanghai cohorts and found seven significant CpG sites related with drug treatment after Bonferroni correction (Figure 4). The CpG sites in CYP3A4 promoter had a methylation rate ranging from 20% to approximately 100%. We also observed that every single CpG site has slight difference between the Henan and Shanghai cohort excepting CYP3A4_CpG_-258 suggesting that the geography factor might have little effect in methylation status during risperidone treatment therapy period in both good and poor responders. The initial two positive CpG sites (CYP3A4_CpG_-36, CYP3A4_CpG_-296, before Bonferroni correction) found in the Henan cohort failed to be validated in Shanghai replication group while another novel positive CpG site (CYP3A4_CpG_-390, P=0.048, P=0.78, before and after Bonferroni correction, respectively) was found. However, in the meta-analysis, CYP3A4_CpG_-36 and CYP3A4_CpG_-296 were validated ultimately and another two novel CpG sites were found (PCYP3A4_CpG_-36=0.0014, PCYP3A4_CpG_-258=0.0013, PCYP3A4_CpG_-296=0.0014, PCYP3A4_CpG_-367:-372:-374=0.028, after Bonferroni correction). Kacevska et al.43 investigated the methylation status in ~12 kb CYP3A4 regulation region including the proximal promoter, XREM and CLEM4 and in separate C/EBP and HNF4α-binding regions in only 79 subjects and found CYP3A4_CpG_-383 showing significant Spearman’s rank coefficient between adjacent CpG sites. Their research covered a wider region of CYP3A4 than ours, but we excavated much more thoroughly in the CYP3A4 promoter region, which contains 16 sites vs 2 sites for analysis. Besides, the four candidate CpG sites were all located before CYP3A4_CpG_-383 and the methylation rates of CYP3A4_CpG_-36, CYP3A4_CpG_-258, CYP3A4_CpG_-296 were higher in poor responders than good responders in both the Henan and Shanghai cohort, possibly suggesting that the inhibition of CYP3A4 protein expression by high methylation rate might result in low efficiency in metabolizing risperidone. Though CYP2D6 enzyme accounts for only 1.3%–4.3% of all hepatic CYPs but metabolites ~20% medications in human liver,44 little knowledge is accessible about methylation regulation in CYP2D6 promoter and gene body regions. Park and colleagues found the methylation frequency in the gene body region of CYP2D6 containing 32 CpG islands, the methylation frequency was 45.5% and 90.3% in human embryonic stem cell-derived hepatocytes and primary hepatocytes, respectively, which exhibits a dynamic methylation pattern change. Our specific CpG sites were not overlapped with that found by Park and colleagues, and we found three positive CYP2D6_CpG sites (PCYP2D6_CpG_193 =0.012, PCYP2D6_CpG_242:244:250 =0.00076, PCYP2D6_CpG_284 =0.034) in meta-analysis in 15 CpG sites. The methylation status of CYP2D6 gene body was highly varied and irregular across CpG site. We also observed that the methylation rate in poor responders was relatively higher than good responders in both the Henan and Shanghai cohort for all the three significant CpG sites, methylation rate of CYP2D6_242:244:250 in poor responders reached 90% which means CYP2D6 enzyme might be repressed and induced undesired risperidone treatment response. Although these findings may still be preliminary and still require either clinical examination or a larger clinical sample group, such translatable pharmacological effects demonstrate the potential of epigenetic phenomenon in explaining the interindividual differences in drug treatment outcome. Characterizing such epigenetic marks and developing noninvasive approaches to examine them holds a promising tool in the effective treatment of schizophrenia.
MDR analysis
Many factors affect risperidone treatment response. Genetic polymorphism or epigenetic regulation alone may be incapable of explaining drug response. A previous study observed this combination effect in drug response.45 In this study, we conducted a combined analysis including all the 27 SNPs and the methylation status of genes that encoded drug metabolism enzyme, drug transporters and neurotransmitter receptors and so on using MDR software. The four-locus model (CYP3A4_CpG_-82:-86+rs6280 (CYP1B1) + rs1800497 (DRD2) + rs6265 (BDNF)) was regarded as the best significant model (P=0.038), which might provide new clues in predicting drug treatment response. Description of the best four-locus model could be found in the Supplementary Materials. This model revealed that mutual interactions of genetic and epigenetic factors might be related with risperidone treatment efficacy in Chinese Han schizophrenia patients.
Conclusion
In conclusion, pharmacogenetics studies of antipsychotic drugs are promising despite many challenges. Our results may push the field closer to routine clinical utilization of pharmacogenetics testing to maximize therapeutic effects and minimize adverse effects. We found genetic and epigenetic biomarkers in risperidone treatment efficacy due to our systematical study design, but there are still some shortcomings in our research and more samples are suggested to be recruited to strengthen our results in future researchs. Progress in genomic technology and bioinformatics, larger sample sizes, better phenotype characterization and precise study design will help to promote antipsychotic pharmacogenetics to its next level.
References
Kane JM . Schizophrenia. N Engl J Med 1996; 334: 34–41.
Arranz MJ, de Leon J . Pharmacogenetics and pharmacogenomics of schizophrenia: a review of last decade of research. Mol Psychiatry 2007; 12: 707–747.
Jovanovic N, Bozina N, Lovric M, Medved V, Jakovljevic M, Peles AM . The role of CYP2D6 and ABCB1 pharmacogenetics in drug-naive patients with first-episode schizophrenia treated with risperidone. Eur J Clin Pharmacol 2010; 66: 1109–1117.
de Leon J, Susce MT, Pan RM, Fairchild M, Koch WH, Wedlund PJ . The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. J Clin Psychiatry 2005; 66: 15–27.
Shinkai T, De Luca V, Utsunomiya K, Sakata S, Inoue Y, Fukunaka Y et al. Functional polymorphism of the human multidrug resistance gene (MDR1) and polydipsia-hyponatremia in schizophrenia. Neuromol Med 2008; 10: 362–367.
Ikeda M, Tomita Y, Mouri A, Koga M, Okochi T, Yoshimura R et al. Identification of novel candidate genes for treatment response to risperidone and susceptibility for schizophrenia: integrated analysis among pharmacogenomics, mouse expression, and genetic case-control association approaches. Biol Psychiatry 2010; 67: 263–269.
Gomez A, Ingelman-Sundberg M . Pharmacoepigenetics: its role in interindividual differences in drug response. Clin Pharmacol Ther 2009; 85: 426–430.
Zhong XB, Leeder JS . Epigenetic regulation of ADME-related genes: focus on drug metabolism and transport. Drug Metab Dispos 2013; 41: 1721–1724.
Ivanov M, Kacevska M, Ingelman-Sundberg M . Epigenomics and interindividual differences in drug response. Clin Pharmacol Ther 2012; 92: 727–736.
Kacevska M, Ivanov M, Ingelman-Sundberg M . Epigenetic-dependent regulation of drug transport and metabolism: an update. Pharmacogenomics 2012; 13: 1373–1385.
Fisel P, Schaeffeler E, Schwab M . DNA Methylation of ADME Genes. Clin Pharmacol Ther 2016; 99: 512–527.
Wei Z, Wang L, Yu T, Wang Y, Sun L, Wang T et al. Histamine H4 receptor polymorphism: a potential predictor of risperidone efficacy. J Clin Psychopharmacol 2013; 33: 221–225.
Shi YY, He L . SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res 2005; 15: 97–98.
Qin S, Zhao X, Pan Y, Liu J, Feng G, Fu J et al. An association study of the N-methyl-D-aspartate receptor NR1 subunit gene (GRIN1) and NR2B subunit gene (GRIN2B) in schizophrenia with universal DNA microarray. Eur J Hum Genet 2005; 13: 807–814.
Mosteller M, Condreay LD, Harris EC, Ambery C, Beerahee M, Ghosh S . Exploring the roles of UGT1A1 and UGT1A3 in oral clearance of GSK2190915, a 5-lipoxygenase-activating protein inhibitor. Pharmacogenet Genomics 2014; 24: 618–621.
Gall WE, Zawada G, Mojarrabi B, Tephly TR, Green MD, Coffman BL et al. Differential glucuronidation of bile acids, androgens and estrogens by human UGT1A3 and 2B7. J Steroid Biochem Mol Biol 1999; 70: 101–108.
Trottier J, El Husseini D, Perreault M, Paquet S, Caron P, Bourassa S et al. The human UGT1A3 enzyme conjugates norursodeoxycholic acid into a C23-ester glucuronide in the liver. J Biol Chem 2010; 285: 1113–1121.
Ramirez J, Ratain MJ, Innocenti F . Uridine 5'-diphospho-glucuronosyltransferase genetic polymorphisms and response to cancer chemotherapy. Future Oncol 2010; 6: 563–585.
Rossi F, Francese M, Iodice RM, Falcone E, Vetrella S, Punzo F et al. [Inherited disorders of bilirubin metabolism]. Minerva Pediatr 2005; 57: 53–63.
Ramirez J, Mirkov S, House LK, Ratain MJ . Glucuronidation of OTS167 in humans is catalyzed by UDP-glucuronosyltransferases UGT1A1, UGT1A3, UGT1A8, and UGT1A10. Drug Metab Dispos 2015; 43: 928–935.
Liu W, Ramirez J, Gamazon ER, Mirkov S, Chen P, Wu K et al. Genetic factors affecting gene transcription and catalytic activity of UDP-glucuronosyltransferases in human liver. Hum Mol Genet 2014; 23: 5558–5569.
Axelrod J, Tomchick R . Enzymatic O-methylation of epinephrine and other catechols. J Biol Chem 1958; 233: 702–705.
Grossman MH, Emanuel BS, Budarf ML . Chromosomal mapping of the human catechol-O-methyltransferase gene to 22q11.1——q11.2. Genomics 1992; 12: 822–825.
Harrison PJ, Weinberger DR . Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 2005; 10: 40–68.
Williams HJ, Owen MJ, O'Donovan MC . Is COMT a susceptibility gene for schizophrenia? Schizophr Bull 2007; 33: 635–641.
Illi A, Mattila KM, Kampman O, Anttila S, Roivas M, Lehtimaki T et al. Catechol-O-methyltransferase and monoamine oxidase A genotypes and drug response to conventional neuroleptics in schizophrenia. J Clin Psychopharmacol 2003; 23: 429–434.
Gupta M, Bhatnagar P, Grover S, Kaur H, Baghel R, Bhasin Y et al. Association studies of catechol-O-methyltransferase (COMT) gene with schizophrenia and response to antipsychotic treatment. Pharmacogenomics 2009; 10: 385–397.
Xu Q, Wu X, Li M, Huang H, Minica C, Yi Z et al. Association studies of genomic variants with treatment response to risperidone, clozapine, quetiapine and chlorpromazine in the Chinese Han population. Pharmacogenomics J 2015; 16: 357–365.
Kleinberg DL, Davis JM, de Coster R, Van Baelen B, Brecher M . Prolactin levels and adverse events in patients treated with risperidone. J Clin Psychopharmacol 1999; 19: 57–61.
Richelson E, Souder T . Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci 2000; 68: 29–39.
Zhao QZ, Liu BC, Zhang J, Wang L, Li XW, Wang Y et al. Association between a COMT polymorphism and clinical response to risperidone treatment: a pharmacogenetic study. Psychiatr Genet 2012; 22: 298–299.
St Clair D, Xu M, Wang P, Yu Y, Fang Y, Zhang F et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959-1961. JAMA 2005; 294: 557–562.
van Os J, Kenis G, Rutten BP . The environment and schizophrenia. Nature 2010; 468: 203–212.
Wang L, Yu L, Zhang AP, Fang C, Du J, Gu NF et al. Serum prolactin levels, plasma risperidone levels, polymorphism of cytochrome P450 2D6 and clinical response in patients with schizophrenia. J Psychopharmacol 2007; 21: 837–842.
Du J, Zhang A, Wang L, Xuan J, Yu L, Che R et al. Relationship between response to risperidone, plasma concentrations of risperidone and CYP3A4 polymorphisms in schizophrenia patients. J Psychopharmacol 2010; 24: 1115–1120.
Huo R, Tang K, Wei Z, Shen L, Xiong Y, Wu X et al. Genetic polymorphisms in CYP2E1: association with schizophrenia susceptibility and risperidone response in the Chinese Han population. PLoS ONE 2012; 7: e34809.
Tang K, Li X, Xing Q, Li W, Feng G, He L et al. Genetic polymorphism analysis of cytochrome P4502E1 (CYP2E1) in Chinese Han populations from four different geographic areas of Mainland China. Genomics 2010; 95: 224–229.
Kim SK, Park HJ, Seok H, Jeon HS, Lee TW, Lee SH et al. Association studies of cytochrome P450, family 2, subfamily E, polypeptide 1 (CYP2E1) gene polymorphisms with acute rejection in kidney transplantation recipients. Clin Transplant 2014; 28: 707–712.
Xing Q, Gao R, Li H, Feng G, Xu M, Duan S et al. Polymorphisms of the ABCB1 gene are associated with the therapeutic response to risperidone in Chinese schizophrenia patients. Pharmacogenomics 2006; 7: 987–993.
Xuan J, Zhao X, He G, Yu L, Wang L, Tang W et al. Effects of the dopamine D3 receptor (DRD3) gene polymorphisms on risperidone response: a pharmacogenetic study. Neuropsychopharmacology 2008; 33: 305–311.
Kacevska M, Ivanov M, Ingelman-Sundberg M . Perspectives on epigenetics and its relevance to adverse drug reactions. Clin Pharmacol Ther 2011; 89: 902–907.
Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13: R43.
Kacevska M, Ivanov M, Wyss A, Kasela S, Milani L, Rane A et al. DNA methylation dynamics in the hepatic CYP3A4 gene promoter. Biochimie 2012; 94: 2338–2344.
Zanger UM, Schwab M . Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013; 138: 103–141.
Ritchie MD, Haas DW, Motsinger AA, Donahue JP, Erdem H, Raffanti S et al. Drug transporter and metabolizing enzyme gene variants and nonnucleoside reverse-transcriptase inhibitor hepatotoxicity. Clin Infect Dis 2006; 43: 779–782.
Acknowledgements
This work was supported by grants from the 863 Program (2012AA02A515, 2012AA021802), the 973 Program (2010CB529600), the National Key Technology R&D Program (2012BAI01B09), the National Nature Science Foundation of China (81421061, 81121001, 81273596, J1210047, 30900799, 30972823), Shanghai Municipal Commission of Science and Technology Program (11DZ1950300, 09DJ1400601), the Public Health Key Disciplines in Shanghai-Health Microbiology (12GWZX0801), Shanghai Key Laboratory of Psychotic Disorders 13dz2260500, Public Science and Technology Research Funds (201210056), the Shanghai Jiaotong University Interdisciplinary Research fund and the Shanghai Leading Academic Discipline Project (B205).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Shi, Y., Li, M., Song, C. et al. Combined study of genetic and epigenetic biomarker risperidone treatment efficacy in Chinese Han schizophrenia patients. Transl Psychiatry 7, e1170 (2017). https://doi.org/10.1038/tp.2017.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.143
This article is cited by
-
Association between the rs6313 polymorphism in the 5-HTR2A gene and the efficacy of antipsychotic drugs
BMC Psychiatry (2023)
-
Persistent and transgenerational effects of risperidone in zebrafish
Environmental Science and Pollution Research (2019)
