
Featured
-
-
Article
| Open Access DOPAnization of tyrosine in α-synuclein by tyrosine hydroxylase leads to the formation of oligomers
DOPAnization of tyrosine in α-synuclein by tyrosine hydroxylase leads to the formation of oligomers
In this work, the authors show that α-synuclein is posttranslationally dopanized at Tyr136 by tyrosine hydroxylase, which facilitates the formation of oligomers. This modification likely impacts pathogenesis and the selective degeneration of dopaminergic neurons in Parkinson’s disease.
- Mingyue Jin
- , Sakiko Matsumoto
- & Shinji Hirotsune
-
Article
| Open Access The 3D structure of lipidic fibrils of α-synuclein
The 3D structure of lipidic fibrils of α-synuclein
Interactions between α-synuclein fibrils and lipids have been associated with the development of Parkinson’s disease. This cryo-EM study reveals structural details of these interactions and suggests a mechanism for fibril-induced lipid extraction.
- Benedikt Frieg
- , Leif Antonschmidt
- & Gunnar F. Schröder
-
Article
| Open Access Cryo-EM structure of an ATTRwt amyloid fibril from systemic non-hereditary transthyretin amyloidosis
Cryo-EM structure of an ATTRwt amyloid fibril from systemic non-hereditary transthyretin amyloidosis
This manuscript reports the structure of a pathologically relevant wild type ATTR amyloid fibril from systemic non-hereditary transthyretin amyloidosis. The comparison of the wild type ATTR fibril with two previous published ex vivo V30M ATTR fibrils highlighted numerous similarities between these different reconstructions, pointing to a common underlying structure for ATTR fibrils despite coming from different mutants and patients.
- Maximilian Steinebrei
- , Juliane Gottwald
- & Matthias Schmidt
-
Article
| Open Access Small soluble α-synuclein aggregates are the toxic species in Parkinson’s disease
Small soluble α-synuclein aggregates are the toxic species in Parkinson’s disease
α-synuclein aggregates cause neuronal damage, but their heterogeneity complicates studying their toxic properties. Here, the authors analyze α-synuclein aggregates in vitro and study post-mortem brain samples, providing evidence that small aggregates are the main culprit for neuronal death in Parkinson’s disease.
- Derya Emin
- , Yu P. Zhang
- & David Klenerman
-
Article
| Open Access The clinical drug candidate anle138b binds in a cavity of lipidic α-synuclein fibrils
The clinical drug candidate anle138b binds in a cavity of lipidic α-synuclein fibrils
Understanding how small molecules bind to pathological aggregates is of importance for therapeutic and diagnostic development in diseases such as Parkinson’s Disease. Here, the authors reveal a binding site of anle138b to lipid-induced α-synuclein fibrils.
- Leif Antonschmidt
- , Dirk Matthes
- & Loren B. Andreas
-
Article
| Open Access Protein import motor complex reacts to mitochondrial misfolding by reducing protein import and activating mitophagy
Protein import motor complex reacts to mitochondrial misfolding by reducing protein import and activating mitophagy
Mitophagy activation is mediated by mitochondrial depolarization. Here, the authors show that mitochondrial protein misfolding can activate mitophagy in a depolarization-independent manner mediated by a protein import reduction.
- Jonas Benjamin Michaelis
- , Melinda Elaine Brunstein
- & Christian Münch
-
Article
| Open Access Designed peptides as nanomolar cross-amyloid inhibitors acting via supramolecular nanofiber co-assembly
Designed peptides as nanomolar cross-amyloid inhibitors acting via supramolecular nanofiber co-assembly
Amyloid self-assembly is linked to type 2 diabetes and Alzheimer’s disease. Here the authors designed constrained peptides which are nanomolar amyloid inhibitors of the key polypeptides IAPP and Aβ42 and act via supramolecular nanofiber co-assembly.
- Karin Taş
- , Beatrice Dalla Volta
- & Aphrodite Kapurniotu
-
Article
| Open Access Single residue modulators of amyloid formation in the N-terminal P1-region of α-synuclein
Single residue modulators of amyloid formation in the N-terminal P1-region of α-synuclein
The authors of this work characterize the effect of amino acid substitution on α-synuclein (α-Syn) aggregation. Residues 38 and 42 (in addition to 39) within the P1 region of α-Syn affect amyloid formation. The effect of substitution at position 38 is dependent on the amino-acid introduced, suggesting that specific interactions control α -Syn aggregation.
- Sabine M. Ulamec
- , Roberto Maya-Martinez
- & David J. Brockwell
-
Article
| Open Access Identification of a HTT-specific binding motif in DNAJB1 essential for suppression and disaggregation of HTT
Identification of a HTT-specific binding motif in DNAJB1 essential for suppression and disaggregation of HTT
Ayala Mariscal et al have identified and characterized the interface of pathogenic Huntingtin and the molecular chaperone DNAJB1. Histidine-244 of the C-terminal domain of DNAJB1 is a key residues for binding to the poly-proline region of HTT. This binding site is specific for the interaction with Huntingtin.
- S. M. Ayala Mariscal
- , M. L. Pigazzini
- & J. Kirstein
-
Article
| Open Access Molecular mechanism for the synchronized electrostatic coacervation and co-aggregation of alpha-synuclein and tau
Molecular mechanism for the synchronized electrostatic coacervation and co-aggregation of alpha-synuclein and tau
Here, the authors report that α-synuclein phase-separates into liquid condensates with positively charged polypeptides such as Tau. The condensates undergo different maturation processes, including the formation of α-synuclein/Tau amyloid hetero-aggregates inside the condensates.
- Pablo Gracia
- , David Polanco
- & Nunilo Cremades
-
Article
| Open Access The Cryo-EM structures of two amphibian antimicrobial cross-β amyloid fibrils
The Cryo-EM structures of two amphibian antimicrobial cross-β amyloid fibrils
In this work the authors provide high-resolution structural support for the amyloid-antimicrobial link via functional amyloids displaying propeller-like and kinked cross-β fibrils.
- Robert Bücker
- , Carolin Seuring
- & Meytal Landau
-
Article
| Open Access Heparin induces α-synuclein to form new fibril polymorphs with attenuated neuropathology
Heparin induces α-synuclein to form new fibril polymorphs with attenuated neuropathology
The Cryo-EM structures reported in this work reveal how heparin incorporates into α-syn fibril formation to determine fibril polymorphs. This highlights the role of biological polymers in the conformational selection and neuropathological regulation of amyloid fibrils.
- Youqi Tao
- , Yunpeng Sun
- & Dan Li
-
Comment
| Open Access The shape of things to come: structural insights into how prion proteins encipher heritable information
The shape of things to come: structural insights into how prion proteins encipher heritable information
The prion hypothesis embodies the radical concept that prion proteins contain the necessary information for infectious replication within their shape, thus obviating the requirement for genomic material. Two elegant papers by Hoyt et al. and Manka et al. describing high-resolution structures of infectious prions bring us closer to answering the long-standing question of how different prion conformations produce heritably distinct diseases.
- Glenn C. Telling
-
Article
| Open Access 2.7 Å cryo-EM structure of ex vivo RML prion fibrils
2.7 Å cryo-EM structure of ex vivo RML prion fibrils
High-resolution structures of mammalian prions have remained elusive. Here, Manka et al. report the cryo-EM structure of infectious RML prion fibrils from mice. Structural similarity with recently reported infectious 263K prion fibrils from hamsters now suggests a common prion architecture.
- Szymon W. Manka
- , Wenjuan Zhang
- & Jonathan D. F. Wadsworth
-
Article
| Open Access Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion
Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion
Misfolded SOD1 has been linked to both familial and sporadic ALS. Here the authors have determined the cryo-EM structure of SOD1 fibrils, providing insights into the conversion of SOD1 from its immature form into an aggregated form during pathogenesis of ALS.
- Li-Qiang Wang
- , Yeyang Ma
- & Yi Liang
-
Article
| Open Access Neurotoxic amyloidogenic peptides in the proteome of SARS-COV2: potential implications for neurological symptoms in COVID-19
Neurotoxic amyloidogenic peptides in the proteome of SARS-COV2: potential implications for neurological symptoms in COVID-19
Here the authors report the formation of toxic clumps of protein, similar to amyloid assemblies found in Alzheimer’s disease and suggest their possible role for some of the neurological symptoms of long-COVID.
- Mirren Charnley
- , Saba Islam
- & Nicholas P. Reynolds
-
Article
| Open Access Fluent molecular mixing of Tau isoforms in Alzheimer’s disease neurofibrillary tangles
Fluent molecular mixing of Tau isoforms in Alzheimer’s disease neurofibrillary tangles
The tau protein in Alzheimer’s disease contains two isoforms. Using solid-state NMR and seeded growth of isotopically labeled tau, here the authors determined that the two isoforms mix fluently on the molecular level to propagate the AD tau structure.
- Aurelio J. Dregni
- , Pu Duan
- & Mei Hong
-
Article
| Open Access Insights into the client protein release mechanism of the ATP-independent chaperone Spy
Insights into the client protein release mechanism of the ATP-independent chaperone Spy
How ATP-independent chaperones release their clients without energy input remains enigmatic. Here the authors discover that chaperone Spy uses its long, disordered N terminus to facilitate client release through competitive, dynamic intramolecular interactions with Spy’s client binding surface.
- Wei He
- , Xinming Li
- & Shu Quan
-
Article
| Open Access Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses
Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses
In this work, the authors report that hyperphosphorylated recombinant tau spontaneously assembles into small, amorphous aggregates, which disrupt membranes and induce Toll-like receptor 4-dependent responses in human macrophages.
- Jonathan X. Meng
- , Yu Zhang
- & David Klenerman
-
Article
| Open Access Stress-induced protein disaggregation in the endoplasmic reticulum catalysed by BiP
Stress-induced protein disaggregation in the endoplasmic reticulum catalysed by BiP
Aggregation of misfolded proteins underlie dementias. Here, the authors show that stressed cells activate an innate mechanism to resolve aggregates of defective proteins in the endoplasmic reticulum, where a third of cellular proteins are produced.
- Eduardo Pinho Melo
- , Tasuku Konno
- & Edward Avezov
-
Article
| Open Access Mapping SP-C co-chaperone binding sites reveals molecular consequences of disease-causing mutations on protein maturation
Mapping SP-C co-chaperone binding sites reveals molecular consequences of disease-causing mutations on protein maturation
Interstitial Lung Disease (ILD)-associated mutations in surfactant protein C (SP-C) render the protein prone to aggregation. Here, the authors reveal their impact on protein maturation, provide insights into recognition of aggregation prone regions by chaperones, and address the autosomal dominant nature of ILD mutants.
- Kristine F. R. Pobre-Piza
- , Melissa J. Mann
- & Linda M. Hendershot
-
Article
| Open Access Structural basis of FPR2 in recognition of Aβ42 and neuroprotection by humanin
Structural basis of FPR2 in recognition of Aβ42 and neuroprotection by humanin
The formyl peptide receptor 2 (FPR2) is involved in the pathogenesis of Alzheimer’s disease. Structures of FPR2 bound to Aβ42, humanin, or formyl peptides offer insight into Aβ42 neurotoxicity, humanin neuroprotection, and FPR ligand selectivity
- Ya Zhu
- , Xiaowen Lin
- & Beili Wu
-
Article
| Open Access Mapping the sequence specificity of heterotypic amyloid interactions enables the identification of aggregation modifiers
Mapping the sequence specificity of heterotypic amyloid interactions enables the identification of aggregation modifiers
In this work, Louros et al. uncover a rule book for interactions of amyloids with other proteins. This grammar was shown to promote cellular spreading of tau aggregates in cells, but can also be harvested to develop structure-based aggregation blockers.
- Nikolaos Louros
- , Meine Ramakers
- & Joost Schymkowitz
-
Article
| Open Access Spatiotemporal modulations in heterotypic condensates of prion and α-synuclein control phase transitions and amyloid conversion
Spatiotemporal modulations in heterotypic condensates of prion and α-synuclein control phase transitions and amyloid conversion
The authors show that prion protein and α-synuclein undergo phase separation through domain-specific electrostatic interactions. These complex coacervates possess electrostatic nanoclusters and can convert into multiphasic condensates and amyloids.
- Aishwarya Agarwal
- , Lisha Arora
- & Samrat Mukhopadhyay
-
Article
| Open Access Differential roles for DNAJ isoforms in HTT-polyQ and FUS aggregation modulation revealed by chaperone screens
Differential roles for DNAJ isoforms in HTT-polyQ and FUS aggregation modulation revealed by chaperone screens
Here, using quantitative screens in human cells, the authors reveal distinct effects of naturally-occurring DNAJ chaperone isoforms on pathological aggregation of the Huntington’s disease-associated HTT-polyQ and the ALS-related mutant FUS.
- Kinneret Rozales
- , Amal Younis
- & Reut Shalgi
-
Article
| Open Access Towards a generic prototyping approach for therapeutically-relevant peptides and proteins in a cell-free translation system
Towards a generic prototyping approach for therapeutically-relevant peptides and proteins in a cell-free translation system
Generic approach for rapid prototyping is essential for the progress of synthetic biology. Here the authors modify the cell-free translation system to control protein aggregation and folding and validate the approach by using single conditions for prototyping of various disulfide-constrained polypeptides.
- Yue Wu
- , Zhenling Cui
- & Sergey Mureev
-
Article
| Open Access Cryo-EM demonstrates the in vitro proliferation of an ex vivo amyloid fibril morphology by seeding
Cryo-EM demonstrates the in vitro proliferation of an ex vivo amyloid fibril morphology by seeding
Here, the authors present the cryo-EM structure of in vitro amyloid fibrils from recombinant SAA1.1 protein that were formed by seeding with fibrils purified from systemic AA amyloidosis tissue. This in vitro fibril structure resembles the structure of the ex vivo fibrils but differs from unseeded in vitro fibrils. These findings show that fibril morphologies can be propagated in vitro by seeding.
- Thomas Heerde
- , Matthies Rennegarbe
- & Marcus Fändrich
-
Article
| Open Access Capillary flow experiments for thermodynamic and kinetic characterization of protein liquid-liquid phase separation
Capillary flow experiments for thermodynamic and kinetic characterization of protein liquid-liquid phase separation
Methods to quantitatively study liquid-liquid phase separation (LLPS) of proteins are lacking. Here the authors report Capillary flow experiments (Capflex) for the quantification of key LLPS parameters; they study Ddx4, the RP3 peptide and the aberrant liquid-to-solid phase transition of α-synuclein.
- Emil G. P. Stender
- , Soumik Ray
- & Alexander K. Buell
-
Article
| Open Access Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties
Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties
The mechanisms underlying Huntingtin protein (Htt) aggregation are not fully understood. Here the authors perform a detailed investigation of the ultrastructural and biochemical properties of huntingtin cytoplasmic and nuclear inclusions, and reveal that they form via distinct mechanisms and exert their toxicity via different pathways.
- Nathan Riguet
- , Anne-Laure Mahul-Mellier
- & Hilal A. Lashuel
-
Article
| Open Access Dissection of the amyloid formation pathway in AL amyloidosis
Dissection of the amyloid formation pathway in AL amyloidosis
AL amyloidosis is caused by the accumulation of overproduced light chain (LC) fragments as fibrils in patient organs and it is the most prevalent systemic amyloidosis. Here, the authors combine biochemical and biophysical experiments to characterise the lag phase of a patient-derived truncated LC and they identify structural transitions that precede fibril formation.
- Pamina Kazman
- , Ramona M. Absmeier
- & Johannes Buchner
-
Article
| Open Access Nascent chains can form co-translational folding intermediates that promote post-translational folding outcomes in a disease-causing protein
Nascent chains can form co-translational folding intermediates that promote post-translational folding outcomes in a disease-causing protein
Alpha-1-antitrypsin (AAT) deficiency results from misfolding-prone AAT variants. Here the authors show that AAT forms co-translational folding intermediates on the ribosome that persist upon release and determine its folding fate. They show too that the ribosome can also modulate misfolding-prone AAT intermediates during their synthesis.
- Elena Plessa
- , Lien P. Chu
- & Lisa D. Cabrita
-
Article
| Open Access Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM
Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM
Systemic AL amyloidosis is caused by misfolding of immunoglobulin light chains (LCs) but how post-translational modifications (PTMs) of LCs influence amyloid formation is not well understood. Here, the authors present the cryo-EM structure of an AL amyloid fibril derived from the heart tissue of a patient that is partially pyroglutamylated, N-glycosylated and contains an intramolecular disulfide bond. Based on their structure and biochemical experiments the authors conclude that the mutational changes, disulfide bond and glycosylation determine the fibril protein fold and that glycosylation protects the fibril core from proteolytic degradation.
- Lynn Radamaker
- , Sara Karimi-Farsijani
- & Marcus Fändrich
-
Article
| Open Access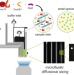 The Hsc70 disaggregation machinery removes monomer units directly from α-synuclein fibril ends
The Hsc70 disaggregation machinery removes monomer units directly from α-synuclein fibril ends
Molecular chaperones from the Hsp70 family can break up protein aggregates, including amyloids. Here, the authors utilize microfluidic diffusional sizing to assess the mechanism of α-synuclein (αS) disaggregation by the Hsc70–DnaJB1–Apg2 system, and show that single αS molecules are removed directly from the fibril ends.
- Matthias M. Schneider
- , Saurabh Gautam
- & Tuomas P. J. Knowles
-
Article
| Open Access DnaJC7 binds natively folded structural elements in tau to inhibit amyloid formation
DnaJC7 binds natively folded structural elements in tau to inhibit amyloid formation
Protein binding by the Hsp70/J-domain protein (JDP) chaperones prevents aggregation of the client protein. Here, the authors show that DnaJC7 binds preferentially to natively folded wild-type tau, via a β-turn element in tau that contains the known amyloid motif, while aggregation-prone tau mutants are recognized with reduced affinity.
- Zhiqiang Hou
- , Pawel M. Wydorski
- & Lukasz A. Joachimiak
-
Article
| Open Access Endo-lysosomal Aβ concentration and pH trigger formation of Aβ oligomers that potently induce Tau missorting
Endo-lysosomal Aβ concentration and pH trigger formation of Aβ oligomers that potently induce Tau missorting
Aβ oligomers (AβO) are thought to represent the main toxic species in Alzheimer’s disease but very high Aβ concentrations are required to study them in vitro and it remains unknown what role these off-pathway oligomers play in vivo. Here, the authors use a dimeric variant of Aβ termed dimAβ, where two Aβ40 units are linked, which facilitates to study AβO formation kinetics and they observe that Aβ off-pathway oligomer formation is strongly accelerated at endo-lysosomal pH, while amyloid fibril formation is delayed. Furthermore, the authors demonstrate that dimAβ is a disease-relevant model construct for pathogenic AβO formation by showing that dimAβ AβOs target dendritic spines and induce AD-like somatodendritic Tau missorting.
- Marie P. Schützmann
- , Filip Hasecke
- & Wolfgang Hoyer
-
Article
| Open Access Huntingtin fibrils with different toxicity, structure, and seeding potential can be interconverted
Huntingtin fibrils with different toxicity, structure, and seeding potential can be interconverted
Huntingtin exon-1 (HTTex1) consists of a N-terminal N17 domain, the disease causing polyQ domain and a C-terminal proline-rich domain (PRD). Here, the authors combine electron paramagnetic resonance (EPR), solid-state NMR with other biophysical method to characterise the structural differences of various HTTex1 fibril types with different toxicity and find that the dynamics and entanglement of the PRD domain differs among them and that the HTTex1 fibrils can be interconverted.
- J. Mario Isas
- , Nitin K. Pandey
- & Ansgar B. Siemer
-
Article
| Open Access Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function
Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function
Amyloid aggregation of mutant p53 contributes to its loss of tumor suppressor function and oncogenic gain-of-function. Here, the authors use a protein mimetic to abrogate mutant p53 aggregation and rescue p53 function, which inhibits cancer cell proliferation in vitro and halts tumor growth in vivo.
- L. Palanikumar
- , Laura Karpauskaite
- & Mazin Magzoub
-
Article
| Open Access α-Helical peptidic scaffolds to target α-synuclein toxic species with nanomolar affinity
α-Helical peptidic scaffolds to target α-synuclein toxic species with nanomolar affinity
α-Synuclein (αS) aggregation is a driver of several neurodegenerative disorders. Here, the authors identify a class of peptides that bind toxic αS oligomers and amyloid fibrils but not monomeric functional protein, and prevent further αS aggregation and associated cell damage.
- Jaime Santos
- , Pablo Gracia
- & Salvador Ventura
-
Article
| Open Access A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells
A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells
Analyzing memory B cell repertoires of the healthy elderly enabled Michalon et al. to develop a recombinant human antibody selective for transthyretin amyloid. This antibody removes cardiac amyloid through recruitment of phagocytic immune cells.
- Aubin Michalon
- , Andreas Hagenbuch
- & Jan Grimm
-
Article
| Open Access Wobble tRNA modification and hydrophilic amino acid patterns dictate protein fate
Wobble tRNA modification and hydrophilic amino acid patterns dictate protein fate
Wobble uridine (U34) tRNA modifications are important for the decoding of AA-ending codons. Here the authors show that while the U34-codon content of mRNAs are predictive of changes in ribosome translation elongation, the resulting outcome in protein expression also relies on specific hydrophilic motifs-dependent protein aggregation and clearance.
- Francesca Rapino
- , Zhaoli Zhou
- & Pierre Close
-
Article
| Open Access GCG inhibits SARS-CoV-2 replication by disrupting the liquid phase condensation of its nucleocapsid protein
GCG inhibits SARS-CoV-2 replication by disrupting the liquid phase condensation of its nucleocapsid protein
Coronavirus nucleocapsid (N) protein is important for viral genome packaging and virion assembly. Here the authors show that natural chemical (-)-gallocatechin gallate (GCG) disrupts the liquid–liquid phase separation of N and inhibits SARS-CoV-2 replication.
- Ming Zhao
- , Yu Yu
- & Tao Li
-
Article
| Open Access UXT chaperone prevents proteotoxicity by acting as an autophagy adaptor for p62-dependent aggrephagy
UXT chaperone prevents proteotoxicity by acting as an autophagy adaptor for p62-dependent aggrephagy
p62/SQSTM1 acts as a key mediator in the selective autophagy of protein aggregates, or aggrephagy. Here the authors identify the prefoldin-like chaperone UXT as an autophagy adaptor of p62 dependent aggrephagy and show that ectopic UXT expression delays motor neuron degeneration in a Xenopus model.
- Min Ji Yoon
- , Boyoon Choi
- & Chungho Kim
-
Article
| Open Access The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells
The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells
The self-assembly of α-synuclein (αS) is a pathological feature of Parkinson’s disease. The αS species responsible for neuronal damage are not well characterized. Here, the authors show that αS fibrils release soluble prefibrillar oligomeric species responsible for neurotoxicity in vitro.
- Roberta Cascella
- , Serene W. Chen
- & Cristina Cecchi
-
Article
| Open Access AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils
AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils
Systemic AA amyloidosis is a protein misfolding disease caused by the formation of amyloid fibrils from serum amyloid A (SAA) protein. Here, the authors present the cryo-EM structures of AA amyloid fibrils isolated from mouse tissue and in vitro formed fibrils, which differ in their structures and they also show that the ex vivo fibrils are more resistant to proteolysis than the in vitro fibrils and propose that pathogenic amyloid fibrils might originate from proteolytic selection.
- Akanksha Bansal
- , Matthias Schmidt
- & Marcus Fändrich
-
Article
| Open Access Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis
Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis
Systemic AL amyloidosis is a protein misfolding disease caused by the aggregation and fibrillation of immunoglobulin light chains (LCs). Here, the authors present the cryo-EM structures of λ3 LC-derived amyloid fibrils that were isolated from patient tissue and they observe structural breaks, where the two different fibril structures co-exist at different z-axial positions within the same fibril.
- Lynn Radamaker
- , Julian Baur
- & Marcus Fändrich
-
Article
| Open Access Infrared nanospectroscopy reveals the molecular interaction fingerprint of an aggregation inhibitor with single Aβ42 oligomers
Infrared nanospectroscopy reveals the molecular interaction fingerprint of an aggregation inhibitor with single Aβ42 oligomers
Our understanding of the molecular mechanisms underlying pathological protein aggregation remains incomplete. Here, single molecule infrared nanospectroscopy (AFM-IR) offers insight into the structure of Aβ42 oligomeric and fibrillar species and their interaction with an aggregation inhibitor, paving the way for single molecule drug discovery studies.
- Francesco Simone Ruggeri
- , Johnny Habchi
- & Tuomas P. J. Knowles
-
Article
| Open Access High-resolution ex vivo NMR spectroscopy of human Z α1-antitrypsin
High-resolution ex vivo NMR spectroscopy of human Z α1-antitrypsin
α1-Antitrypsin (AAT) is a 52 kDa serum glycoprotein, the misfolding and polymerisation of which is associated with COPD and liver disease. Here the authors demonstrate the use of high-resolution multidimensional solution-state NMR spectroscopy to characterise the structure and dynamics in solution of Z AAT purified directly from clinical patients.
- Alistair M. Jagger
- , Christopher A. Waudby
- & David A. Lomas
-
Article
| Open Access Sis1 potentiates the stress response to protein aggregation and elevated temperature
Sis1 potentiates the stress response to protein aggregation and elevated temperature
Identifying factors that enable cells to induce a potent stress response to amyloid-like aggregation can provide further insight into the mechanism of stress regulation. Here, the authors express polyglutamine-expanded Huntingtin as a model disease protein in yeast cells and perform a genetic screen for chaperone factors that allow yeast cells to activate a potent stress response. They identify Sis1, an essential Hsp40 co-chaperone of Hsp70, as a critical sensor of proteotoxic stress and further show that both Sis1 and its mammalian homolog DnaJB6 regulate the magnitude of the cellular heat stress response, indicating that this mechanism is conserved.
- Courtney L. Klaips
- , Michael H. M. Gropp
- & F. Ulrich Hartl
-
Article
| Open Access TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease
TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease
TDP-43 inclusions are observed in Alzheimer’s disease. Here the authors show that TDP-43 interacts with amyloid-β and inhibits fibrillization in vitro and exacerbates Alzheimer’s disease pathology in animal models.
- Yao-Hsiang Shih
- , Ling-Hsien Tu
- & Yun-Ru Chen
 Cryo-EM structure of ex vivo fibrils associated with extreme AA amyloidosis prevalence in a cat shelter
Cryo-EM structure of ex vivo fibrils associated with extreme AA amyloidosis prevalence in a cat shelter