
Article
|
Open Access
Featured
-
-
Article
| Open Access GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs
GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs
Structural variants may be omitted in sequence analysis despite their importance in genome variation and phenotypic impact. Here the authors present GraphTyper2, which uses pangenome graphs to genotype structural variants using short-reads and can be applied in large-scale sequencing studies.
- Hannes P. Eggertsson
- , Snaedis Kristmundsdottir
- & Pall Melsted
-
Article
| Open Access Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads
Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads
Repetitive sequences in complex eukaryote genomes can cause fragmented assemblies with incomplete gene sequences and unanchored or mispositioned contigs. Here, the authors report HERA, a method to improve genome assemblies by efficiently resolving repeats using single-molecule sequencing data.
- Huilong Du
- & Chengzhi Liang
-
Article
| Open Access CUTseq is a versatile method for preparing multiplexed DNA sequencing libraries from low-input samples
CUTseq is a versatile method for preparing multiplexed DNA sequencing libraries from low-input samples
Genomics DNA library preparation from formalin-fixed paraffin-embedded tissues is challenging. Here the authors describe CUTseq that uses restriction enzymes and in vitro amplification to barcode samples for reduced representation genome sequencing.
- Xiaolu Zhang
- , Silvano Garnerone
- & Nicola Crosetto
-
Article
| Open Access Linked-read sequencing of gametes allows efficient genome-wide analysis of meiotic recombination
Linked-read sequencing of gametes allows efficient genome-wide analysis of meiotic recombination
Meiotic crossovers (COs) generate genetic variation and ensure proper chromosome segregation. Here, the authors develop a method for identifying COs at kilobase resolution in pooled recombinants using linked-read sequencing data, and apply it to investigate genome-wide CO landscapes of Arabidopsis thaliana.
- Hequan Sun
- , Beth A. Rowan
- & Korbinian Schneeberger
-
Article
| Open Access Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses
Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses
Despite the importance of grapevine cultivation in human history and the economic values of cultivar improvement, large-scale genomic variation data are lacking. Here the authors resequence 472 Vitis accessions and use the identified genetic variations for domestication history, demography, and GWAS analyses.
- Zhenchang Liang
- , Shengchang Duan
- & Yang Dong
-
Article
| Open Access Genome-wide profiling of adenine base editor specificity by EndoV-seq
Genome-wide profiling of adenine base editor specificity by EndoV-seq
Adenine base editors are an important contribution to the genome editing toolbox. Here the authors present EndoV-seq, an endonuclease-based assay for evaluating genomewide off-target effects of base editing.
- Puping Liang
- , Xiaowei Xie
- & Zhou Songyang
-
Article
| Open Access Robust single-cell DNA methylome profiling with snmC-seq2
Robust single-cell DNA methylome profiling with snmC-seq2
Single-cell DNA methylome profiling allows the study of epigenomic heterogeneity in tissues but has been impeded by library quality. Here the authors demonstrate snmC-seq2 which improves mapping, throughput and library complexity.
- Chongyuan Luo
- , Angeline Rivkin
- & Joseph R. Ecker
-
Article
| Open Access Nanoplanktonic diatoms are globally overlooked but play a role in spring blooms and carbon export
Nanoplanktonic diatoms are globally overlooked but play a role in spring blooms and carbon export
Diatoms are major oceanic primary producers, but some species belonging to the nano- and even picoplankton size are poorly characterized. Here the authors describe a massive spring bloom of the smallest known diatom in the Mediterranean Sea and reveal their general oversight at the global scale.
- Karine Leblanc
- , Bernard Quéguiner
- & Pascal Conan
-
Article
| Open Access High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell
High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell
Long-read sequencing technologies facilitate efficient and high quality genome assembly. Here Michael et al. achieve a fast reference assembly for Arabidopsis thaliana KBS-Mac-74 accession using the handheld Oxford Nanopore MinION sequencer and consumer computing hardware, and demonstrate its usefulness in resolving complex structural variation.
- Todd P. Michael
- , Florian Jupe
- & Joseph R. Ecker
-
Article
| Open Access Extreme haplotype variation in the desiccation-tolerant clubmoss Selaginella lepidophylla
Extreme haplotype variation in the desiccation-tolerant clubmoss Selaginella lepidophylla
Selaginella lepidophylla is a clubmoss with extreme desiccation tolerance. Here, the authors assemble its highly heterozygotic haplotypes and examine gene expression changes during desiccation, which shed light on the mechanisms for maintaining a small genome size and adaptation to extreme drying.
- Robert VanBuren
- , Ching Man Wai
- & Todd P. Michael
-
Article
| Open Access Dense and accurate whole-chromosome haplotyping of individual genomes
Dense and accurate whole-chromosome haplotyping of individual genomes
Haplotype information is important in investigating many biological phenomena. Here, Porubsky et al. combine Strand-seq with long-read or linked-read sequencing to obtain complete and genome-wide haplotypes of a single individual genome at manageable costs.
- David Porubsky
- , Shilpa Garg
- & Tobias Marschall
-
Article
| Open Access Single nucleus sequencing reveals spermatid chromosome fragmentation as a possible cause of maize haploid induction
Single nucleus sequencing reveals spermatid chromosome fragmentation as a possible cause of maize haploid induction
Plant breeders can produce haploid maize lines using haploid inducer lines as pollen donors. Here, by sequencing the genomes of single pollen nuclei, Li et al. show that haploid inducer spermatids are frequently aneuploid and suggest chromosome fragmentation as a possible cause of haploid induction.
- Xiang Li
- , Dexuan Meng
- & Jianbing Yan
-
Article
| Open Access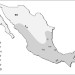 Demographic history and biologically relevant genetic variation of Native Mexicans inferred from whole-genome sequencing
Demographic history and biologically relevant genetic variation of Native Mexicans inferred from whole-genome sequencing
People of Mexico have diverse historical and genetic background. Here, Romero-Hidalgo and colleagues sequence whole genomes of Native Americans of Mexico, and show demographic history and genetic variation shared among subgroups of Native Americans.
- Sandra Romero-Hidalgo
- , Adrián Ochoa-Leyva
- & Xavier Soberón
-
Article
| Open Access Whole genome analysis of a schistosomiasis-transmitting freshwater snail
Whole genome analysis of a schistosomiasis-transmitting freshwater snail
Biomphalaria glabrata is a fresh water snail that acts as a host for trematode Schistosoma mansoni that causes intestinal infection in human. This work describes the genome and transcriptome analyses from 12 different tissues of B glabrata, and identify genes for snail behavior and evolution.
- Coen M. Adema
- , LaDeana W. Hillier
- & Richard K. Wilson
-
Article
| Open Access CRISPR–Cas9-targeted fragmentation and selective sequencing enable massively parallel microsatellite analysis
CRISPR–Cas9-targeted fragmentation and selective sequencing enable massively parallel microsatellite analysis
Microsatellite genotyping is a common molecular method of for identification but current methods have restricted throughput. The authors demonstrate STR-Seq, which combines next generation sequencing with targetedin vitroCRISPR-Cas9 fragmentation to enable massively parallel analysis in the thousands.
- GiWon Shin
- , Susan M. Grimes
- & Hanlee P. Ji
-
Article
| Open Access TruePrime is a novel method for whole-genome amplification from single cells based on TthPrimPol
TruePrime is a novel method for whole-genome amplification from single cells based on TthPrimPol
Single cell genomic analysis needs DNA amplification with high fidelity and accuracy. Here, the authors devise a novel multiple displacement amplification method called TruePrime that is based in Thermus thermophilusPrimPol and Phi29 DNA polymerase, and demonstrate its utility and accuracy.
- Ángel J. Picher
- , Bettina Budeus
- & Armin Schneider
-
Article
| Open Access A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome
A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome
The Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA) aims to better understand population genetics of the African diaspora. Here, it uses deeply sequenced whole-genomes to describe the impact of admixture and potential disease burden of deleterious variants.
- Rasika Ann Mathias
- , Margaret A. Taub
- & Kathleen C. Barnes
-
Article
| Open Access Droplet barcoding for massively parallel single-molecule deep sequencing
Droplet barcoding for massively parallel single-molecule deep sequencing
The ability to accurately sequence long DNA molecules is important across biology. Here, Lan et al. report a droplet-based method that barcodes single DNA molecules, allowing the full-length molecules to be sequenced with multi-fold coverage using short-read next-generation sequencing.
- Freeman Lan
- , John R. Haliburton
- & Adam R. Abate
-
Article
| Open Access Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer
Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer
The genomic features of metastatic prostate cancer are beginning to be understood. Here, the authors performed whole genome sequencing of plasma samples from these patients and found a high plasticity of the cancer genomes with newly occurring focal amplifications as a driving force in progression.
- Peter Ulz
- , Jelena Belic
- & Michael R. Speicher
-
Article
| Open Access Characterization of eukaryotic DNA N6-methyladenine by a highly sensitive restriction enzyme-assisted sequencing
Characterization of eukaryotic DNA N6-methyladenine by a highly sensitive restriction enzyme-assisted sequencing
DNA N6-methyladenine is prevalent in prokaryotes, and is recently also detected in eukaryotes such as roundworm and fly. Here, Luo et al. report a DpnI-assisted base resolution method that detects 6mA genome-wide with nanograms of input DNA and lower sequencing depth than the previous restriction enzyme-based approach.
- Guan-Zheng Luo
- , Fang Wang
- & Chuan He
-
Article
| Open Access Fast and sensitive mapping of nanopore sequencing reads with GraphMap
Fast and sensitive mapping of nanopore sequencing reads with GraphMap
Read mapping and alignment tools are critical for many applications based on MinION sequencers. Here, the authors present GraphMap, a mapping algorithm designed to analyze nanopore sequencing reads, that progressively refines candidate alignments to handle potentially high error rates to align long reads.
- Ivan Sović
- , Mile Šikić
- & Niranjan Nagarajan
-
Article
| Open Access A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing
A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing
Cancer genetics has benefited from the advent of next generation sequencing, yet a comparison of sequencing and analysis techniques is lacking. Here, the authors sequence a normal-tumour pair and perform data analysis at multiple institutes and highlight some of the pitfalls associated with the different methods.
- Tyler S. Alioto
- , Ivo Buchhalter
- & Ivo G. Gut
-
Article
| Open Access Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency
Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency
Osteosarcomas are a heterogenous group of tumours and little is known about how these tumours evolve. Here, Kovac et al. use exome sequencing and discover that although no driver gene explains the majority of these tumours, they are characterized by specific mutation signatures and genomic instability typical of BRCA1/2-deficient tumours.
- Michal Kovac
- , Claudia Blattmann
- & Daniel Baumhoer
-
Article
| Open Access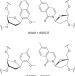 Identification of DNA lesions using a third base pair for amplification and nanopore sequencing
Identification of DNA lesions using a third base pair for amplification and nanopore sequencing
Genomic DNA lesions exist in low levels and cannot be amplified by standard PCR. Here, Riedlet al. report a method to amplify damaged DNA sites by replacing them via DNA repair with unnatural base pairs, which are subsequently identified by Sanger sequencing or α-hemolysin nanopore sequencing.
- Jan Riedl
- , Yun Ding
- & Cynthia J. Burrows
-
Article
| Open Access Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals
Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals
The Tohoku Medical Megabank Organization establishes a biobank with detailed patient health care and genome information. Here the authors analyse whole-genome sequences of 1,070 Japanese individuals, allowing them to catalogue 21 million single-nucleotide variants including 12 million novel ones.
- Masao Nagasaki
- , Jun Yasuda
- & Masayuki Yamamoto
-
Article
| Open Access Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes
Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes
Bacterial DNA methylation is involved in many processes, from host defense to antibiotic resistance, however current methods for examining methylated genomes lack single-cell resolution. Here Beaulaurier et al. present Single Molecule Modification Analysis of Long Reads, a new tool for de novodetection of epigenetic heterogeneity.
- John Beaulaurier
- , Xue-Song Zhang
- & Gang Fang
-
Article
| Open Access TCF12 is mutated in anaplastic oligodendroglioma
TCF12 is mutated in anaplastic oligodendroglioma
Anaplastic oligodendrogliomas are rare and incurable primary brain tumours with few treatment options. Here Labrecheet al. perform whole-exome sequencing and identify recurring mutations in transcription factor TCF12, which are associated with aggressive tumours.
- Karim Labreche
- , Iva Simeonova
- & Michel Wager
-
Article
| Open Access Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling
Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling
Gene transfer is a powerful technique to investigate the mechanistic basis of tumorigenesis. Here Zuckermann et al. adapt CRISPR/Cas9 genome editing to target potential oncogenes somatically in vivo, establishing a fast and convenient system for validating novel genetic candidates.
- Marc Zuckermann
- , Volker Hovestadt
- & Jan Gronych
-
Article
| Open Access Phasing of single DNA molecules by massively parallel barcoding
Phasing of single DNA molecules by massively parallel barcoding
DNA phasing information — the determination of which specific sequences belong to the same DNA molecule—is not easily obtained from sequencing applications that rely on short reads. Here the authors develop a phasing method based on massively parallel barcoding of single DNA molecules.
- Erik Borgström
- , David Redin
- & Afshin Ahmadian
-
Article |
 Calibrating genomic and allelic coverage bias in single-cell sequencing
Calibrating genomic and allelic coverage bias in single-cell sequencing
Artifacts caused by whole-genome amplification bias are a recurrent challenge in single-cell sequencing analysis. Here, the authors develop statistical models and demonstrate an efficient strategy for controlling amplification errors by a joint analysis of single cell genomes.
- Cheng-Zhong Zhang
- , Viktor A. Adalsteinsson
- & J. Christopher Love
-
Article
| Open Access Increased prevalence of EPAS1 variant in cattle with high-altitude pulmonary hypertension
Increased prevalence of EPAS1 variant in cattle with high-altitude pulmonary hypertension
Pulmonary hypertension and congestive right heart failure afflict some cattle living at high altitude in an autosomal dominant pattern, yet no responsible genes have been identified. Here Newman et al.use whole-exome sequencing to identify variants in the hypoxia inducible factor gene, EPAS1.
- John H. Newman
- , Timothy N. Holt
- & Rizwan Hamid
-
Article |
 High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq
High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq
Characterizing mammalian gene expression regulation by enhancer elements is complicated by the size and complexity of the genome. Here Vanhille et al.demonstrate CapStarr-Seq, a novel high-throughput method for assessing potential enhancers and deciphering the mechanisms regulating transcription
- Laurent Vanhille
- , Aurélien Griffon
- & Salvatore Spicuglia
-
Article |
 Whole-exome sequencing implicates UBE3D in age-related macular degeneration in East Asian populations
Whole-exome sequencing implicates UBE3D in age-related macular degeneration in East Asian populations
Age-related macular degeneration is a prominent cause of irreversible blindness among the elderly. Here Huang et al.identify a novel missense variant in UBE3D that sheds new light on the pathogenesis of the disease.
- Lv-Zhen Huang
- , Ying-Jie Li
- & Xiao-Xin Li
-
Article
| Open Access Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets
Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets
Diagnosis of pancreatic ductal adenocarcinoma (PDA) has poor long-term survival rates with limited therapy options. Here Witkiewicz et al.use microdissection and whole-exome sequencing to identify novel recurrent PDA mutations, highlighting the genetic diversity of this aggressive cancer.
- Agnieszka K. Witkiewicz
- , Elizabeth A. McMillan
- & Erik S. Knudsen
-
Article |
 Metal ion-directed dynamic splicing of DNA through global conformational change by intramolecular complexation
Metal ion-directed dynamic splicing of DNA through global conformational change by intramolecular complexation
Higher-order structured DNA molecules can be manipulated to carry out specific enzymatic functions. Here the authors demonstrate the metal ion-directed global conformational control of DNA structure, using intramolecular coordination chemistry to manipulate the DNAzyme activity.
- Toshihiro Ihara
- , Hiroyuki Ohura
- & Yusuke Kitamura
-
Article
| Open Access Dissecting meiotic recombination based on tetrad analysis by single-microspore sequencing in maize
Dissecting meiotic recombination based on tetrad analysis by single-microspore sequencing in maize
The crossovers and gene conversions that occur during meiotic recombination contribute to genome diversity in eukaryotes. Here Li et al. describe a method of isolating individual microspores for whole-genome sequencing, providing new insights into the generation of genome diversity through sexual reproduction.
- Xiang Li
- , Lin Li
- & Jianbing Yan
-
Article |
 Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value
Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value
Triple-negative breast cancers (TNBCs) are a heterogeneous group of cancers with varying prognoses. Here, the authors carry out whole-genome methylation capture sequencing from TNBC samples and matched normal samples, and identify differentially methylated regions that define a potentially novel TNBC signature.
- Clare Stirzaker
- , Elena Zotenko
- & Susan J. Clark
-
Article
| Open Access DNA sequencing using polymerase substrate-binding kinetics
DNA sequencing using polymerase substrate-binding kinetics
Next-generation sequencing technologies vary in performance, which is often measured by metrics such as sequencing speed, accuracy and read length. Here, the authors present a new sequencing by synthesis method that monitors polymerase binding to DNA, and suggest that this method has the potential to generate longer and faster reads.
- Michael John Robert Previte
- , Chunhong Zhou
- & Molly Min He
-
Article
| Open Access Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours
Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours
Testicular germ cell tumour (TGCT) is the most common cancer in young men. Here, the authors sequence the whole exomes of 42 TGCTs, and characterize the mutational profile of this tumour type.
- Kevin Litchfield
- , Brenda Summersgill
- & Clare Turnbull
-
Article
| Open Access An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins
An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins
Type 2 diabetes (T2D) is a highly heterogeneous disease with a strong genetic component. Here the authors examine genome-wide methylation patterns in T2D-discordant, T2D-concordant and healthy concordant monozygotic twin pairs, and identify DNA methylation signals that may represent new biomarkers or drug targets for T2D.
- Wei Yuan
- , Yudong Xia
- & Tim D. Spector
-
Article
| Open Access Identification of the remains of King Richard III
Identification of the remains of King Richard III
King Richard III was a controversial English King whose remains are presumably deposited in Grey Friars in Leicester. Here the authors sequence the mitochondrial genome and Y-chromosome DNA of the skeletal remains and living relatives of Richard III and confirm that the remains belong to King Richard III.
- Turi E. King
- , Gloria Gonzalez Fortes
- & Kevin Schürer
-
Article
| Open Access Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts
Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts
Plasmodium falciparum, known to cause malaria in humans, evolved from parasites of African Great Apes. Here, the authors compare the genome of the human parasite, P. falciparum, with those of two related chimpanzee parasites, P. reichenowi and P. gaboni, and provide insight into the genetic basis of P. falciparumadaptation to human hosts.
- Thomas D. Otto
- , Julian C. Rayner
- & Matthew Berriman
-
Article |
 Sequencing-based approach identified three new susceptibility loci for psoriasis
Sequencing-based approach identified three new susceptibility loci for psoriasis
Although psoriasis is a chronic disorder affecting approximately 2% of the population, little is known about the underlying genetic architecture. Here, the authors carry out exome sequencing in a large Han Chinese cohort of psoriasis patients and healthy controls, and identify three new genes that may increase risk of developing the disease.
- Yujun Sheng
- , Xin Jin
- & Xuejun Zhang
-
Article |
 Semiconductor-based DNA sequencing of histone modification states
Semiconductor-based DNA sequencing of histone modification states
Semiconductor-based, non-optical DNA sequencing technologies such as Ion Torrent sequencing offer speed and cost advantages compared with alternative techniques. Cheng et al. demonstrate a protocol allowing the use of Ion Torrent technology to sequence DNA from chromatin immunoprecipitation experiments.
- Christine S. Cheng
- , Kunal Rai
- & Ido Amit
-
Article
| Open Access Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution
Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution
The wild rice species can be used as germplasm resources for this crop’s genetic improvement. Here Chen and colleagues report the de novo sequencing of the O. brachyanthagenome, and identify the origin of genome size variation, the role of gene movement and its implications on heterochromatin evolution in the rice genome.
- Jinfeng Chen
- , Quanfei Huang
- & Mingsheng Chen
-
Article
| Open Access Genome sequences of wild and domestic bactrian camels
Genome sequences of wild and domestic bactrian camels
Camels are essential means of transport in deserts, but we know little about the biology of these extraordinary mammals. This study reports the genome sequences of the wild and domestic bactrian camel, offering a glimpse into the camels’ genetic adaptation to harsh environments.
- Jirimutu
- , Zhen Wang
- & He Meng
-
Article
| Open Access Genome sequence of the model medicinal mushroom Ganoderma lucidum
Genome sequence of the model medicinal mushroom Ganoderma lucidum
Ganoderma lucidumis a macrofungus in traditional Chinese medicine known to produce different bioactive compounds. In this study, the genome ofG. lucidumis sequenced, making this organism a potential model system for future studies of secondary metabolic pathways and their regulation in medicinal fungi.
- Shilin Chen
- , Jiang Xu
- & Chao Sun
-
Article |
 Discovery of lost diversity of paternal horse lineages using ancient DNA
Discovery of lost diversity of paternal horse lineages using ancient DNA
Modern female horses are genetically diverse but male horses are relatively homogenous. Lippoldet al. sequence the Y chromosome of nine ancient horses and detect diversity in the ancestral paternal lineage, demonstrating ancient Y-chromosomal DNA sequencing can provide insights into evolution.
- Sebastian Lippold
- , Michael Knapp
- & Michael Hofreiter
 Accurate, scalable and integrative haplotype estimation
Accurate, scalable and integrative haplotype estimation