


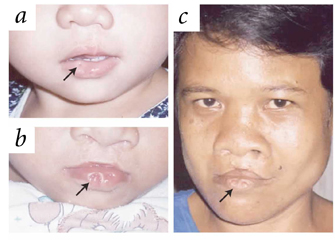
 Figure 1: Van der Woude syndrome
Figure 1: Van der Woude syndrome














« Prev Next »
Researchers have made dramatic inroads into the study of polygenic and other complex human diseases, due in large part to knowledge of the human genome sequence, the generation of widespread markers of genetic variation, and the development of new technologies that allow investigators to associate disease phenotypes with genetic loci. Although polygenic diseases are more common than single-gene disorders, studies of monogenic diseases provide an invaluable opportunity to learn about underlying molecular mechanisms, thereby contributing a great deal to our understanding of all forms of genetic disease.
Mendel Revisited: Monogenic Diseases
The human genome contains an estimated total of 20,000-25,000 genes that serve as blueprints for building all of our proteins (International Human Genome Sequencing Consortium, 2004). In single-gene diseases, a mutation in just one of these genes is responsible for disease. Single-gene diseases run in families and can be dominant or recessive, and autosomal or sex-linked. Pedigree analyses of large families with many affected members are very useful for determining the inheritance pattern of single-gene diseases. Table 1 includes some examples of single-gene diseases.
Table 1: Examples of Human Diseases, Modes of Inheritance, and Associated Genes
OMIM, Online Mendelian Inheritance in Man, is a regularly updated, online database established in 1997 by Dr. Victor A. McKusick that is focused on inherited genetic diseases in humans. As of June 15, 2008,* OMIM reported 387 human genes of known sequence with a known phenotype, and 2,310 human phenotypes with a known molecular basis. However, OMIM also reported 1,621 confirmed Mendelian phenotypes for which the molecular basis is not known. Furthermore, OMIM reported 2,084 phenotypes for which a Mendelian basis is suspected but has not been fully established, or that may exhibit overlap with other characterized phenotypes. As you can see, more questions than answers remain regarding the identity of single genes and their role in human disease.
Trends in Gene Discovery
After the human genome was sequenced, researchers began to shift their focus from monogenic diseases to polygenic diseases, which involve many genes. There are several reasons for this movement toward polygenic diseases. For one, many of the 1,621 monogenic disorders without known genes are very rare. As a result, researchers face difficulties in identifying families with the disease and in obtaining sufficient numbers of DNA samples for comparison to unaffected family members. Also, funding agencies, biotechnology companies, and pharmaceutical companies are often less likely to invest financial resources in research efforts focused on rare diseases.
However, studies of monogenic diseases contribute a great deal to knowledge of polygenic forms of human disease (Antonarakis & Beckmann, 2006). To this end, monogenic diseases are most worthy of our attention.
Back to the Future: Using New Technologies to Find Old Genes
Before the human genome was sequenced, researchers relied on labor-intensive, slow-going techniques for mapping and isolating disease-associated genes. For instance, although efforts to isolate the gene associated with Huntington's disease began in the late 1970s, the gene was not identified until 1993.
With the sequence of the human genome available, researchers have been able to generate maps of each chromosome, showing the precise location of every gene and determining areas of the genome that can differ from one person to the next (termed "polymorphic"). Of special interest to researchers are single nucleotide polymorphisms (SNPs), which are single base pair polymorphic regions. SNPs occur throughout the human genome at an average rate of one SNP per every 1,000 base pairs. The International HapMap Project has mapped SNPs along the length of every human chromosome and has made this information freely available to scientists worldwide. These polymorphic SNP markers can be used to map disease-associated genes.
Researchers have developed methods for the simultaneous analysis of up to one million different SNPs throughout the entire human genome using genomic DNA isolated from a blood sample (or any other biological source of DNA) and a single DNA chip, which is a small silicon glass wafer onto which single-stranded DNA fragments can be adhered in a grid-like pattern. A SNP chip contains short, single-stranded DNA molecules called oligonucleotides that correspond to known SNP variants. The DNA isolated from the blood sample is broken into fragments, labeled with a fluorescent dye, and converted into single-stranded DNA. The single-stranded, fluorescently labeled genomic DNA fragments are then incubated with the SNP chip, and only those DNA fragments that are a perfect match will bind to their complementary SNP oligonucleotide on the SNP chip grid. A laser is then used to scan each grid position to determine which SNP variants are represented.
By using SNP chips, researchers can obtain a SNP profile of an individual that spans the entire genome. SNP profiles can be compared between affected and unaffected family members (or unaffected, unrelated individuals) to determine which SNPs segregate with a disease (or are associated with the disease). Because the SNP sequences have already been mapped to specific chromosomal locations, researchers can also immediately map the disease-associated gene to a specific region of a given human chromosome.
The Bioinformatics Era: Genomics and Proteomics
Bioinformatics is the genome-inspired field of biology that analyzes genomic information to predict gene and protein function. Bioinformaticists can easily examine a region of a chromosome and determine which segments correspond to protein-encoding genes. Furthermore, they can compare the sequence of a gene of unknown function to the rest of the genome and find similar genes with known functions. Based on similarity between genes, researchers can often predict how gene-encoded proteins may function within a cell.
Large-scale studies of genes and proteins are referred to as genomics and proteomics, respectively. Researchers can now use gene chips to simultaneously examine the expression (mRNA) levels of all human protein-encoding genes from a given cell population. Gene chips are similar in concept to SNP chips, but their grids contain single-stranded DNA fragments that correspond to protein-encoding genes. In order to study gene expression, researchers first isolate mRNA from a tissue of interest, then convert it into single-stranded complementary DNA (cDNA) and label it with a fluorescent dye. The single-stranded, fluorescently labeled cDNA is then incubated with the gene chip, allowing hybridization between the cDNA molecules and their complementary sequences on the gene chip grid. A laser is used to scan the chip and determine the fluorescent signal associated with every mRNA represented on the gene chip grid system to yield a gene expression profile for a given individual. By comparing gene expression profiles from normal and diseased individuals, scientists can also determine changes in gene expression associated with human disease.
More recently, researchers have developed chip-based methods for the simultaneous examination of thousands of proteins within a given cell population. In these approaches, the protein chip grid contains adhered antibodies that recognize and bind to specific human proteins. The protein chip is incubated with a fluorescently labeled protein sample from a given individual, and a laser is used to scan the chip to determine the levels of each protein represented by the antibodies on the grid. In this way, researchers can determine the proteomic profile associated with a given form of human disease, and they can see which proteins show altered expression.
Databases
As you can imagine, genomic and proteomic approaches, which simultaneously examine thousands of genes and proteins, generate a tremendous amount of data. Furthermore, scientists are publishing new data at a very fast pace. In order to make meaningful connections among worldwide scientific discoveries, a number of databases have been established. Examples of useful databases include OMIM and Entrez Gene, which provide a number of useful links to other databases.
From Man to Mouse: Using Genetic Model Organisms to Understand Single-Gene Diseases in Humans
Due to the remarkable level of homology between genomes across the evolutionary tree, scientists can learn a lot about the underlying molecular mechanisms associated with single-gene diseases in humans by studying organisms that are much simpler: mice, frogs, worms, flies, and even yeast. Many of the genes found in humans are also present in these other types of organisms. Moreover, several of the same basic cellular processes are shared among humans and these organisms, including metabolism, cell division, growth regulation, and more. Although this discussion focuses on mouse models, many seminal discoveries relevant to our understanding of human disease have come from studies of the same type of yeast used to make bread and beer.
Similar to that of humans, the entire sequence of the mouse genome is known. Many human genes are also found in mice, and using mice as a model organism for genetic studies has contributed to our understanding of human disease. Today, researchers can generate mice with a mutation or deletion of a disease-associated gene. They can carry out detailed phenotypic analyses of the mutant mice and learn how the corresponding gene may function in humans. For example, researchers have developed a mouse model of Huntington's disease, in which the mutant mice carry the expanded CAG repeat within the Huntington's disease-associated gene. Although genetically engineered mice are not perfect models of human disease, they can offer valuable insights into the function of disease-associated genes.
© 2002 Courtesy of Jeffrey C. Murray, MD, University of Iowa. All rights reserved. 
Research regarding single-gene human diseases has also uncovered "modifier" genes that can alter the severity of phenotypes associated with mutations in the primary disease-associated gene. For instance, for many years, cystic fibrosis was considered a single-gene disease associated with mutations in the cystic fibrosis-associated gene, CFTR. However, the initial discovery of the CFTR gene was followed by the identification of several additional genes that contribute to cystic fibrosis; several modifier genes have also been identified that can modulate the phenotypes associated with mutations in CFTR (Guggino & Stanton, 2006). Cystic fibrosis-associated phenotypes due to mutations in the CFTR gene are in turn modulated by mutations in the following genes: gastrointestinal phenotypes (MUC1), pulmonary phenotypes (TNF, TGFB1, and MBL2), bowel obstruction at birth/meconium ileus (CFM1), and microbial infections (NOS1).
In addition, studies of monogenic disease transmission in identical twins have uncovered various nongenetic mechanisms associated with disease. For example, identical twins with the same mutation in the gene associated with Duchenne muscular dystrophy, called DMD, can exhibit strikingly different disease phenotypes due to different patterns of X chromosome inactivation (Abbadi et al., 1994).
Finally, monogenic syndromes can sometimes serve as models for complex diseases. Consider the example of Van der Woude syndrome, which is characterized by lower lip pits, orofacial clefts, and even occasional hypodontia. This disorder is caused by dominant mutations in the IRF6 (interferon regulatory factor 6) gene (Kondo et al., 2002). Scientists have proposed that IRF6 variation may also contribute to isolated cleft lip with or without cleft palate (Zucchero et al., 2004), a complex birth defect suggested to be caused by as many as three to 14 genes (Schliekelman & Slatkin, 2002). Indeed, studies with a large sample data set have shown that IRF6 variation contributes to an expressive proportion of isolated cleft lip with or without cleft palate cases (Zucchero et al., 2004). In related studies, researchers isolated tooth agenesis, another complex phenotype commonly found in the general population and that is present in a subset of cases of Van der Woude syndrome, and they showed that IRF6 variation contributes to this condition as well (Vieira et al., 2007). Such results are of interest because they indicate that the same gene can cause a disease as rare as Van der Woude syndrome (with a frequency of 1:100,000 births; Figure 1) and also contribute to much more common defects, such as isolated cleft lip with or without cleft palate (frequency of 1:700 births) and isolated tooth agenesis (frequency of 1:100 births), that have more complex genetic etiologies.
From Simple Beginnings to Complex Endings
Armed with knowledge of the human genome sequence and an arsenal of new molecular tools for gene discovery, today's gene hunters are prepared to greatly expand our knowledge of disease-associated genes. Most certainly, our collective knowledge of single-gene diseases, with the help of databases and reference systems, has the potential to advance our understanding of all types of human disease in ways far greater than imagined at the time of each individual discovery.
* Current statistics from OMIM and other sources can be found on the ClinVar website. As of March 19, 2015, OMIM reported 3,358 genes with a phenotype-causing mutation, and 5,434 phenotypes with a known molecular basis (http://omim.org/statistics/geneMap).
References and Recommended Reading
Abbadi, N., et al. Additional case of female monozygotic twins discordant for the clinical manifestations of Duchenne muscular dystrophy due to opposite X-chromosome inactivation. American Journal of Medical Genetics 52, 198–206 (1994)
Antonarakis, S. E., & Beckmann, J. S. Mendelian disorders deserve more attention. Nature Reviews Genetics 7, 277–282 (2006) doi10.1038/nrg1826 (link to article)
Badano, J. L., & Katsanis, N. Beyond Mendel: An evolving view of human genetic disease transmission. Nature Reviews Genetics 3, 779–789 (2002) doi: 10.1038/nrg910
Fraga, M. F., et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences 102, 10604–10609 (2005)
Guggino, W. B., & Stanton, B. A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nature Reviews Molecular Cell Biology 7, 426–436 (2006) doi:1038/nrm1949 (link to article)
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004) doi:10.1038/nature03001 (link to article)
Kondo, S., et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nature Genetics 32, 285–289 (2002) doi:10.1038/ng985 (link to article)
Muenke, M. The pit, the cleft and the web. Nature Genetics 32, 219–220 (2002) doi:10.1038/ng1002-219 (link to article)
Schliekelman, P., & Slatkin, M. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. American Journal of Human Genetics 71, 1369–1385 (2002)
Vieira, A. R., et al. Interferon regulatory factor 6 (IRF6) and fibroblast growth factor receptor 1 (FGFR1) contribute to human tooth agenesis. American Journal of Medical Genetics 143, 538–545 (2007)
Zucchero, T. M., et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. New England Journal of Medicine 351, 769–780 (2004)










