


















« Prev Next »
In the 1940s, J. B. S. Haldane observed that many red blood cell disorders, such as sickle-cell anemia and various thalassemias, were prominent in tropical regions where malaria was endemic (Haldane, 1949; Figure 1). Haldane hypothesized that these disorders had become common in these regions because natural selection had acted to increase the prevalence of traits that protect individuals from malaria. Just a few years later, Haldane's so-called "malaria hypothesis" was confirmed by researcher A. C. Allison, who demonstrated that the geographical distribution of the sickle-cell mutation in the beta hemoglobin gene (HBB) was limited to Africa and correlated with malaria endemicity. Allison further noted that individuals who carried the sickle-cell trait were resistant to malaria (Allison, 1954).
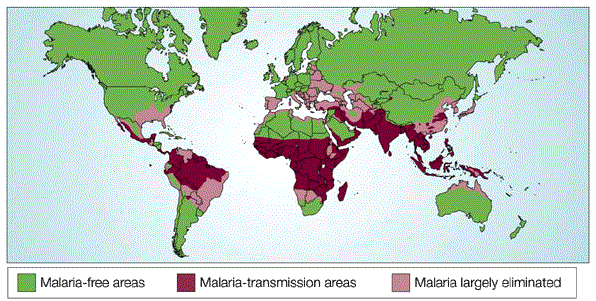
Figure 1: Worldwide distribution of malaria
The worldwide distribution of Plasmodium falciparum malaria in 2003.
© 2003 Modified with permission from the World Health Organization. All rights reserved. 
Allison's confirmation of Haldane's hypothesis provided the first elucidated example of human adaptation since natural selection had been proposed a century earlier. Today, this and other demonstrations of natural selection help point researchers toward biological mechanisms of resistance to infectious disease. Moreover, such examples also shed light on the ways in which pathogens rapidly evolve to remain agents of human morbidity and mortality.
Selection for Malaria Resistance: A Closer Look
Since Allison and Haldane's work, the action of natural selection on genetic resistance to malaria has been shown in a multitude of contexts (Kwiatkowski, 2005). Indeed, the sickle-cell variant (i.e., the HbS allele) has been identified in four distinct genetic backgrounds in different African populations, suggesting that the same mutation arose independently several times through convergent evolution. Beyond HbS, other distinct mutations in the HBB gene have generated the HbC and HbE alleles, which arose and spread in Africa and in Southeast Asia, respectively.
The various HBB alleles aren't alone in offering protection against malaria, however. The geographic distributions of several other red blood cell disorders, including a-thalassemia, G6PD deficiency, and ovalocytosis, correlate to malaria endemicity, and the diseases also are linked to malaria resistance. An even more striking worldwide geographical difference exists for a mutation in the Duffy antigen gene (FY), which encodes a membrane protein used by the Plasmodium vivax malaria parasite to enter red blood cells. This mutation disrupts the protein, thus conferring protection against P. vivax malaria, and it occurs at a prevalence of 100% throughout most of sub-Saharan Africa yet is virtually absent outside of Africa. Moreover, through convergent evolution, an independent mutation in FY that decreases this gene's expression has also become prevalent in Southeast Asia.
So, why has malaria exerted such strong selective pressure? Scientists now know the answer. Malaria is arguably one of the human population's oldest diseases and greatest causes of morbidity and mortality. Research indicates that the malaria-causing parasite Plasmodium falciparum has occurred in human populations for approximately 100,000 years, with a large population expansion in the last 10,000 years as human populations began to move into settlements (Hartl, 2004). P. falciparum, together with the other malaria species, P. vivax, P. malariae, and P. ovale, infects hundreds of millions of people worldwide each year, and kills more than 1 million children annually (World Health Organization, 2000). Because this disease is so devastating, humans have had to evolve adaptive traits to survive in the face of this infectious condition over the past few millennia (Kwiatkowski, 2005).
Broader Implications of Natural Selection for Investigating Infectious Disease
While malaria is the best-understood example of an infectious disease that has driven human evolution, numerous other infectious diseases have also acted in human populations over generations, thus allowing resistance alleles to emerge and spread over time (Diamond, 2005). Based on historical records from the last millennium, these diseases might include smallpox in ancient Europe and in Native American populations, as well as cholera, tuberculosis, and bubonic plague in Europe. Many diseases in Africa have likely been endemic for even longer, such as numerous diarrheal diseases, yellow fever, and Lassa hemorrhagic fever.
Today, with access to heretofore unprecedented data sets for the study of human genetic variation, researchers can exploit the genetic signatures of natural selection using novel analytical methods. In this way, they can identify genetic variants conferring resistance to infectious diseases that have spread through human populations over time. These studies will help elucidate natural mechanisms of defense and perhaps uncover novel evolutionary pressures. Moreover, the same tools that have revolutionized the study of natural selection in humans will also make unprecedented studies of pathogens possible.
Investigating the signatures of natural selection can help elucidate the evolutionary adaptations that have allowed humans to withstand some of our most complex and challenging selective agents. In particular, researchers can look for variants that might be readily detected in genetic association studies; for distinctive, detectable patterns of genetic variation in the human genome; and for clues as to how pathogens themselves evolve so rapidly.
Searching for Variants via Association Studies
By driving highly protective variants to high prevalence, natural selection produces variants that might be readily detected in genetic association studies to help elucidate the biological basis of disease resistance. The classic examples of host genetic factors that play a role in resistance to malaria, such as HbS, are some of the strongest and most robust signals of genetic susceptibility to infectious disease (Hill, 2006). This is because natural selection acts to increase the prevalence of highly advantageous alleles, over time generating common resistance alleles of especially strong effect. For example, a study of genetic susceptibility of HbS in the Gambia detected a significant level of protection using just 315 cases and 583 controls (Ackerman et al., 2005). By studying other ancient selective pressures in which common resistance alleles of strong effect are acting, scientists may have the power to detect a genetic association even with small sample sizes.
In contrast, no single highly protective variant for emergent diseases like HIV and tuberculosis (in Africa) would have had time to spread. For these diseases, resistance appears to be modulated by many rare genetic variants, most with modest protective effect, and genetic studies require extremely large sample sizes (Hill, 2006). This is likely not a biological but, rather, a historical difference. Indeed, hundreds of structural and regulatory mutations exist in HBB, such as HbS, HbE, or HbC, but in populations under malaria selective pressure, a single highly protective variant will often dominate (Kwiatkowski, 2005). Moreover, many variants nearby on the chromosome will rise in prevalence in the population through genetic hitchhiking, such that other nearby linked alleles can serve as proxies for the underlying causal allele in genetic association studies, further enhancing researchers' ability to detect an association. Thus, natural selection may produce important genetic resistance loci that can more easily be detected in association studies.
Searching for Patterns of Variation
As genetic variants conferring resistance to infectious diseases spread through human populations over time through natural selection, they leave distinctive, detectable patterns of genetic variation in the human genome. These signals of selection can uncover novel resistance alleles or even novel evolutionary pressures. Also, as previously mentioned, as advantageous alleles under positive selection rise in prevalence, variants at nearby locations on the same chromosome (linked alleles) also rise in prevalence. Such genetic hitchhiking leads to a "selective sweep" that alters the typical pattern of genetic variation in the region. Selective sweeps produce numerous detectable signals of selection (Nielsen, 2005; Sabeti et al., 2006). As tests for selection have been applied to newly available genetic variation data across the human genome, many of the top signals of selection that have been identified have been at genes and alleles known to be involved with malaria susceptibility, including HBB, FY, CD36, and HLA. These signals were identified in just 90 individuals randomly chosen from the population, and they could have been identified without prior knowledge of a specific variant or selective advantage.
Surveys of natural selection can not only identify new resistance variants for known selective pressures, but they can also potentially uncover previously unrecognized selective pressures. For example, in a genome survey of the Yoruba people of Nigeria, two of the top signals of selection were at genes (LARGE and DMD) biologically linked to the Lassa hemorrhagic fever virus (Sabeti et al., 2007). While little studied, Lassa virus in fact infects many millions of West Africans, and based on oral records and epidemiology, it is likely to be an ancient disease (Richmond & Baglole, 2003). Researchers have documented that in several affected West African populations, between 50% and 90% of individuals are resistant to the virus, suggesting that protective alleles emerged at some point (McCormick & Fisher-Hoch, 2002). This finding could open new avenues for research and shine light on other important pathogens in human history.
Searching for Clues about Pathogen Evolution
The same tools that revolutionized the study of natural selection in humans are now making unprecedented studies of pathogens possible, allowing scientists to better understand how these organisms rapidly evolve to remain agents of human morbidity and mortality. Pathogens are perhaps the most intriguing of all the forces shaping humans. They have had a tremendous impact on our evolution, and they, themselves, evolve over time. The great effect that pathogens have exerted on the human genome is demonstrated by positive selection for traits such as sickle-cell hemoglobin (Sabeti et al., 2006). Natural human defenses have similarly exerted strong pressures on the genomes of pathogens, as has the use of drugs and vaccines (Volkman et al., 2007). By studying genetic diversity in pathogens, researchers can examine how they have evolved to avoid human immune defenses and therapeutics. Furthermore, scientists can investigate in real time the evolutionary consequences of new vaccines and drugs, with the goal of developing better intervention strategies.
Future Endeavors
Investigation of the links between natural selection and disease resistance has revealed some of the forces that have shaped our species, and the findings of these studies have direct implications for human health. However, research thus far represents just a first glimpse of a vast new landscape. In the years to come, new technologies and analytic methods will enable researchers to learn even more about the genetic basis of evolutionary adaptations that have allowed humans to withstand a wide variety of complex and challenging selective agents.
References and Recommended Reading
Ackerman, H., et al. A comparison of case-control and family-based association methods: The example of sickle-cell and malaria. Annals of Human Genetics 69, 559–565 (2005)
Allison, A. C. Protection afforded by sickle-cell trait against subtertian malarial infection. British Medical Journal 4857, 290–294 (1954)
Diamond, J. M. Guns, Germs, and Steel: The Fates of Human Societies (New York, Norton, 2005)
Haldane, J. B. S. Disease and evolution. Ricerca Science Supplement 19, 3–10 (1949)
Hartl, D. L. The origin of malaria: Mixed messages from genetic diversity. Nature Reviews Microbiology 2, 15–22 (2004) doi:10.1038/nrmicro795 (link to article)
Hill, A. V. Aspects of genetic susceptibility to human infectious diseases. Annual Review of Genetics 40, 469–486 (2006)
Kwiatkowski, D. P. How malaria has affected the human genome and what human genetics can teach us about malaria. American Journal of Human Genetics 77, 171–192 (2005)
McCormick, J. B., & Fisher-Hoch, S. P. Lassa fever. Current Topics in Microbiology and Immunology 262, 75–109 (2002)
Nielsen, R. Molecular signatures of natural selection. Annual Review of Genetics 39, 197–218 (2005)
Richmond, J. K., & Baglole, D. J. Lassa fever: Epidemiology, clinical features, and social consequences. British Medical Journal 327, 1271–1275 (2003)
Sabeti, P. C., et al. Positive natural selection in the human lineage. Science 312, 1614–1620 (2006) doi:10.1126/science.1124309
———. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007) (link to article)
Volkman, S. K., et al. A genome-wide map of diversity in Plasmodium falciparum. Nature Genetics 39, 113–119 (2007) doi:10.1038/ng1930 (link to article)
World Health Organization. WHO expert committee on malaria. World Health Organization Technical Report Series 892, 1–74 (2000)










