


 Figure 1: Platforms for proteomics and functional genomics
Figure 1: Platforms for proteomics and functional genomics















« Prev Next »
Great things are done by a series of small things brought together.
—Vincent Van Gogh
One of the major milestones of the budding branch of science known as genomics was the decoding of the human genome. More than 3 gigabases of DNA sequence found their way to the databases and into scientific publications. Yet scarcely a decade later, biologists have progressed beyond sequencing genomes as a major goal. We want to know what organizes genomes in different organisms, and we want to tinker until we have organisms with the smallest genome possible. How does everything fit in one big interacting network of genes, mRNA, and proteins? Recently, scientists have made steps towards answering this question.
Why Was Decoding the Human Genome So Important?
So far, the principal accomplishment of the field of genomics must surely be considered the unravelling of the DNA sequence of the human genome (Lander et al. 2001, Venter et al. 2001). Few scientific feats have received a better press (Ouzounis & Mazière 2006): deciphering the human gene code has been compared to the decoding of the Rosetta Stone (Petsko 2001), the exploration of space (Vukmirovic & Tilghman 2000), and the discovery of nuclear fission (Schrader 2001). Of course, it was in many ways a remarkable milestone in the development of science. Unravelling the human genome first and foremost provided us with a road map for the study of human genetics, helping us to discover new genes and new gene functions. Secondly, the decoding of the genome turned out to be an intriguing competition (and in the final stages, collaboration) between a consortium of world-wide government-paid scientists on the one hand and the private company Celera, owned by J. Craig Venter. Thirdly, the project created a need for fast and reliable sequencing technology. At the start in 1990, science was completely relying on Sanger's dideoxy method (at a very slow speed, and a cost of $1 per base pair sequenced). But by the time the human genome was finished, and a first diploid version of an single individual human's genome (again, that of J. Craig Venter) was ready to be published (Levy et al. 2007), this cost had fallen by a factor of 1000, due to a series of new methods, such as 454 pyrosequencing, Illumina sequencing or Polony sequencing (Hall 2007, Morozova & Marra 2008). And while even the attempt to sequence Venter's genome was still performed using the older Sanger method (Sanger et al. 1977), it was followed in 2008 with a complete reading of the genome of James Watson, sequenced by way of parallel, 454 sequencing technology (Ronaghi et al. 1998, Margulies et al. 2005, Wheeler et al. 2008). Prices and analysis times are predicted to fall even more, with the appearance of even more modern sequencing technologies like Helicos or fluorescence resonance energy transfer (FRET)-based single-molecule sequencing systems, or nanoknife edge probes (Blow 2008), down to $1000 (Mardis 2006), and maybe even to $100 for an entire human sequence (Singer 2008).
With these fast sequencing technologies increasingly available, more and larger genomes could be sequenced, leading to cross-species genome comparisons. These comparisons are meant to find common regulatory sequences, gene conservation and patterns of conservation in noncoding sequences (Ureta-Vidal et al. 2003, Bernardi 2008).
This table gives a non-exhaustive taste of the genomes that have been completed to date. These genomes come from all different branches on the Tree of Life, providing a number of landmarks for future molecular research. Examples of current research already include:
- the restoration of the mammoth genome through comparison of isolated mammoth sequences with those of extant family members such as the African elephant (Poinar et al. 2006);
- the comparison of the Neanderthal genome with that of modern man (Burbane et al. 2010, Noonan 2010);
- the identification of C-box binding factors for enhanced freezing and drought tolerance in plants (Gutterson & Zhang 2004) or of cell division regulating genes (Vandepoele et al. 2002) leading to the identification of target genes for gene modification in plants;
- reconstruction of the evolution of the large primates (and the position of Homo sapiens within this group) (Hacia 2001).
The list is endless, and every new paper comes up with exciting insights to add to the previous ones.
Beyond Sequencing: Time for In-Depth Analysis of the Data
Apart from looking at differences in genome structure and composition between two species, attempts are being made to sequence a number of individuals from the same species. Studies like that will give us the opportunity to assess the variation in the genetic make-up between members of the same species. Obviously, the human species is one of the species being investigated in this manner. Researchers hope to find clues towards, among other things, personalised medical treatment for different ailments (Leary et al. 2010).
Another question that is being asked, concerns the so-called "minimal genome": what is the minimum complement of genes needed to sustain an independent, free living organism? And if you take this one step further — if you know what is minimally needed to make a cell work — it is possible to create such an organism from scratch (recently shown by Gibson et al. 2010), and to add all desired qualities needed for a certain biotechnological task, and not a bit more. Mankind will then be able to create designer organisms, ready to perform any job we need to see done (Szostak et al. 2001, Patil et al. 2004).
The World of Microbes
Genomics has also found a much-needed microbiological application: metagenomics (Handelsman 2004). Until recently, microbiologists had to rely on their ability to make pure cultures of single micro-organisms to be able to identify them, first using biochemical and physiological methods (outlined in Bergey's Manual of Determinative Microbiology); later, with the aid of PCR and simple Sanger sequencing, the base-pair sequence of the 16S rRNA gene of the bacteria in pure culture could be determined (Woese & Fox 1977, Giovannoni et al. 1990), which could then serve as a kind of marker for the position of the bacterium in the taxonomy of the prokaryotes (see the evaluation of the method by Case et al. 2007). Nowadays, using the massive potential that has been unlocked, we are able to extract all DNA from a certain ecological sample (a soil or water sample, a biofilm), break this DNA into pieces, and sequence everything using high throughput methods (Handelsman 2004, Medini et al. 2008). Probably the most famous example is the expedition of the Global Ocean Sampling expedition aboard the Sorcerer II, headed by J. Craig Venter (Rusch et al. 2007). The ship collected seawater samples and performed shotgun sequencing (i.e., breaking down all DNA present in the sample and sequencing it in a massive parallel sequencing effort). To quote from the abstract by Venter et al. (2004):
A total of 1.045 billion base pairs of nonredundant sequence was generated, annotated, and analyzed to elucidate the gene content, diversity, and relative abundance of the organisms within these environmental samples. These data are estimated to derive from at least 1800 genomic species . . . We have identified over 1.2 million previously unknown genes represented in these samples, including more than 782 new rhodopsin-like photoreceptors.
The major challenge in this strategy is, evidently, the amount of puzzle work that is required to cluster and order the DNA fragments that belong to the same organism. We can therefore start to identify many organisms that belong to a certain ecosystem about whose existence we had no clue at all before. Again, this has led to a number of interesting research projects which might have important applications for medicine or food production. For example, scientists have started to analyse and map the "human microbiome", the community of micro-organisms associated with the human body (Ley et al. 2006a, Kuczynski et al. 2010). Knowing more about how mankind lives in symbiosis with certain microbes helps us to better understand (and deal with) diseases like obesity (Ley et al. 2006b, Tschöp et al. 2009, Sekirov et al. 2010).
Some would say that genomics has been able to distil some humility into humankind. The finalised version of the human genome deprived us of the illusion that we are one of the most complex creatures on Earth — an illusion that was at the basis of some guesses that Homo sapiens was expected to have at least 100,000 genes. When we look at a table of genomes by species, and specifically at the number of genes that have been counted or estimated for each species, we notice that humans are surpassed by several plants and invertebrates.
Studying Gene Expression from 35,000 Feet
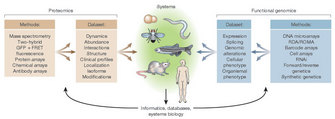
© 2003 Nature Publishing Group Tyers, M. & Mann, M. From genomics to proteomics. Nature 422, 193-197 (2003). All rights reserved. 
To study gene expression differences between two sets of cells (tissues, organisms), we isolate the mRNA from each set. Either group of mRNAs is labeled with a fluorescent dye, then both groups are pipetted onto the surface of the array. On that array, you can find small spots (around 100 μm in diameter) consisting of oligonucleotide strands of about 60 bases long, which is typically representative of one gene within the organism under study. If more mRNA for a certain gene is labelled with one fluorescent label than with the other (because that gene is expressed more), there will be more label attached to the oligonucleotide spot on the array surface. Using lasers to excite the fluorescent labels, we are able to quantify how much less or more mRNA was available in either set of cells. Gene expression becomes quantifiable in this manner, and therefore amenable to statistics (Churchill 2002, Cui & Churchill 2003, Leung & Cavalieri 2003) (Figure 1).
Armed with this powerful technique, scientists have found themselves able to answer a variety of questions like: what happens in a cell when it transforms itself into a metastasizing tumour cell (Klein & Hölze 2006, Leary et al. 2010)? What is the molecular cause of neurological disorders (Niculescu & Kelsoe 2001)? How does a plant respond to an overdose of ozone in the air (Matsuyama et al. 2002), or a chronic dosage of UV-B (Hectors et al. 2007)? How does an organism respond to a toxic component in its environment, and can that response be quantified through a genome-wide analysis of its gene expression patterns (Pennie et al. 2001, Lettieri 2006, Scholin 2010)? And the possibilities are still endless, including the identification of specific disease subforms, identification of novel genes, the annotation of functions to genes, among many others. (Stoughton 2005).
The Complexities of the Transcriptome and the Proteome
Of course, knowing about the abundance of the different mRNAs in a cell is not enough to be able to fully comprehend the complex metabolism of a cell (as defended at length by Anderson & Anderson 1998). In brief — as every biochemistry textbook will explain in detail — gene products (such as proteins) interact with one another in such a way that their activity can be modulated through the action of other proteins (Krueger & Srivastava 2006). Proteins called kinases and phosphorylases activate or deactivate each other by covalently binding phosphate groups on other proteins — for example, in the regulation of the cell cycle or apoptosis (King & Cidlowski 1998). Signal transduction, DNA repair and immunity thrive on proteins being phosphorylated and dephosphorylated as well (Deribe et al. 2010). Other types of these post-translational (i.e., directly on the protein) modifications are: acetylations, nitrosylations and ribosylations (Clark et al. 2005). All these modifications, however poorly understood to date, are bound to have an impact on the activity of a protein, and hence, they are able to alter a cell's metabolism. They may therefore also be very important aspects of determining whether a cell becomes cancerous (Krueger & Srivastava 2006).
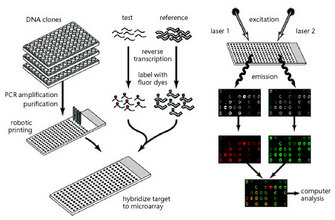
© 1999 Nature Publishing Group Duggan, D. J. et al. Expression profiling using cDNA microarrays. Nature Genetics 21, 10-14 (1999) doi:10.1038/4434. All rights reserved.
Because of the posttranslational modifications and the different splicing variants, the proteome is even more complex to analyse than the transcriptome or the genome. Proteome analysis is also rather labor-intensive and requires large and expensive equipment for running 2D-gels and performing mass spectrometry, whereas a lab that only wants to use microarrays, needs a specific laser scanner at most. Therefore, and to provide a full view of what happens in a cell, the transcriptome and the proteome approaches are bound to work closely together (Figure 2). Specifically, different methods are available for proteome analysis (Rabilloud 2002; Görg et al. 2004). Some are based on 2D gel electrophoresis, others on a combination of 2D gel electrophoresis with mass spectrometry, or on protein arrays. For protein arrays, a given surface will contain a number of protein-recognizing molecules such as antibodies (Jenkins & Pennington 2001; Templin et al. 2003; Gevaert & Vandekerckhove 2000).
"Omics" Biology Is Expanding
The overview of the different aspects of systems biology is far from complete, and probably won't be for a long time. As is normally the case in a new field of science, Systems Biology is going through a phase where a lot of new insights and ways to explore them are popping up. Strikingly, these aspects are unified in their respective names, all ending in "-omics" (Lederberg & McCray 2001). Apart from genomics, transcriptomics and proteomics, we now talk about metabolomics (the study of all the metabolites in a cell, and how they interact — Fiehn 2001) and the subdisciplines lipidomics, antioxidomics or glycomics (dealing with, respectively, lipids, antioxidants and sugars). There is also ecotoxicogenomics, which is using functional genomics to study the effect of toxic substances on living organisms. With a more focused lens, there is phosphoproteomics, the study of all proteins bearing regulatory phosphate groups, as well as kinomics, the selected study of all kinases. And extending beyond cell biology, there are the fledgling fields of mechanomics (all mechanical subsystems in an organism), speechomics (the study of human speech patterns) and bibliomics (the collection of scientific data). Will there one day be a scientomics? Certainly that's going a bit too far.
Summary
Dissecting all the interactions between DNA, mRNA and proteins is an arduous task. However, in the words of 17th century Dutch poets, "nil volentibus arduum" — for those that want, nothing is hard. Grasping the interactions between all these components in a living cell is at the heart of what cell biology and molecular biology want to accomplish. The first steps towards that aim have been made, and scientists have carved out niches for themselves by defining many "omics". Joining all these omics at the root is the desire to understand the systems, the interconnected mechanisms behind the study object. That in itself it the logical continuation of science itself. Given this daunting task, however, we are sure to enjoy the path towards its completion for a very long time to come.
References and Recommended Reading
Anderson, N. L. & Anderson N. G. Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis 19, 1853–1861 (1998). doi: 10.1002/elps.1150191103
Baker, B. S. Sex in flies: the splice of life. Nature 340, 521–524 (1989). doi: 10.1038/340521a0.
Bernardi, G. Fish genomics: A mini-review on some structural and evolutionary issues. Marine Genomics 1, 3–7 (2008). doi: 10.1016/j.margen.2008.04.004
Black, D. L. Mechanisms of alternative pre-messenger RNA splicing. Annual Reviews of Biochemistry 72, 291–336 (2003). doi: 10.1146/annurev.biochem.72.121801.161720.
Blow, N. DNA sequencing: generation next-next. Nature Methods 5, 267–274 (2008). doi: 10.1038/nmeth0308-267
Burbane H. A., Hodges E. et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science 328, 723–725 (2010). doi: 10.1126/science.1188046
Case, R. J., Boucher, Y. et al. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Applied and Environmental Microbiology 73, 278–288 (2007). doi: 10.1128/AEM.01177-06.
Churchill. G. A. Fundamentals of experimental design for cDNA microarrays. Nature Genetics 32, 490–495 (2002). doi: 10.1038/ng1031.
Clark, R. S .B., Bayir, H. et al. Posttranslational protein modifications. Critical Care Medicine 33, S407–409 (2005). doi: 10.1097/01.CCM.0000191712.96336.51
Cui, X. & Churchill, G.A. Statistical tests for differential expression in cDNA microarray experiments. Genome Biology 4, 210 (2003).
Deribe, Y. L., Pawson, T. et al. Post-translational modifications in signal integration. Nature Structural & Molecular Biology 17, 666–672 (2010). doi: 10.1038/nsmb.1842.
Duggan, D. J., Bittner, M. et al. Expression profiling using cDNA microarrays. Nature Genetics 21, 10–14 (1999).
Fiehn, O. Combining genomics, metabolome analysis,and biochemical modelling to understand metabolic networks. Comparative and Functional Genomics 2, 155–168 (2001). doi: 10.1002/cfg.82.
Gevaert, K. & Vandekerckhove, J. Protein identification methods in proteomics. Electrophoresis 21, 1145–1154 (2000). doi: 10.1002/(SICI)1522-2683(20000401)21:6<1145::AID-ELPS1145>3.0.CO;2-Z
Gibson, D. G., Glass, J. I. et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56 (2010). doi: 10.1126/science.1190719
Gilbert, W. Why genes in pieces? Nature 271, 501 (1978).
Giovannoni, S. J., Britschgi, T. B. et al. Genetic diversity in Sargasso Sea bacterioplankton. Nature 345, 60–63 (1990). doi: 10.1038/345060a0
Görg, A., Weiss, W. et al. Current two-dimensional electrophoresis technology for proteomics. Proteomics 4, 3665–3685 (2004). doi: 10.1002/pmic.200401031
Gutterson, N. & Zhang J. Z. Genomics applications to biotech traits: a revolution in progress? Current Opinion in Plant Biology 7, 226–230 (2004). doi: 10.1016/j.pbi.2003.12.002
Hacia, J. G. Genome of the apes. Trends in Genetics 17, 637–645 (2001). doi: 10.1016/S0168-9525(01)02494-5
Hall, N. Advanced sequencing technologies and their wider impact in microbiology. Journal of Experimental Biology 209, 1518–1525 (2007). doi: 10.1242/jeb.001370.
Handelsman, J. Metagenomics: Application of Genomics to Uncultured Microorganisms. Microbiology and Molecular Biology Reviews 68, 669–685 (2004). doi: 10.1128/MMBR.69.1.195.2005.
Hardiman, G. Microarray platforms — comparisons and contrasts. Pharmacogenomics 5, 487–502 (2004).
Hectors, K., Prinsen, E. et al. Arabidopsis thaliana plants acclimated to low dose rates of ultraviolet B radiation show specific changes in morphology and gene expression in the absence of stress symptoms. New Phytologist 175, 255–270 (2007). doi: 10.1111/j.1469-8137.2007.02092.x
Jenkins, R. E. & Pennington, S. R. Arrays for protein expression profiling: towards a viable alternative to two-dimensional gel electrophoresis? Proteomics 1, 13–29 (2001).
King, K. L. & Cidlowski, J. A. Cell cycle regulation and apoptosis. Annual Review of Physiology 60, 601–617 (1998).
Klein, C. A. & Hölzel, D. Systemic cancer progression and tumor dormancy: mathematical models meet single cell genomics. Cell Cycle 5, 1788–1798 (2006).
Krueger, K. E. & Srivastava, S. Posttranslational Protein Modifications. Current Implications for Cancer Detection, Prevention, and Therapeutics. Molecular & Cellular Proteomics 5, 1799–1810 (2006). doi: 10.1074/mcp.R600009-MCP200.
Kuczynski, J., Costello, E. K. et al. Direct sequencing of the human microbiome readily reveals community differences. Genome Biology 11, 210 (2010).
Lander, E. S., Linton, L. M. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–892 (2001). doi: 10.1038/35057062
Leary, R. J., Kinde, I. et al. Development of personalized tumor biomarkers using massively parallel sequencing. Science Translational Medicine 2, 20ra14 (2010). doi: 10.1126/scitranslmed.3000702
Lederberg, J. & McCray, A. T. 'Ome Sweet 'Omics — A Genealogical Treasury of Words. The Scientist 15, 8 (2001).
Lettieri, T. Recent Applications of DNA microarray technology to toxicology and ecotoxicology. Environmental Health Perspective 114, 4–9 (2006). doi: 10.1289/ehp.8194
Leung, Y. F. & Cavalieri, D. Fundamentals of cDNA microarray data analysis. Trends in Genetics 19, 649–659 (2003). doi: 10.1016/j.tig.2003.09.015
Levy, S., Sutton, G. et al. The diploid genome sequence of an individual human. Public Library of Science, Biology 5, e254 (2007). doi: 10.1371/journal.pbio.0050254
Ley, R. E. & Peterson, D. A. et al. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848 (2006a). doi: 10.1016/j.cell.2006.02.017
Ley, R. E., Turnbaugh, P. J. et al. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006b). doi: 10.1038/4441022a
Mardis, E. R. Anticipating the $1,000 genome. Genome Biology 7, 112 (2006). doi: 10.1186/gb-2006-7-7-112
Margulies, M., Egholm, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 326–327 (2005). doi: 10.1038/437326a
Matlin, A. J., Clark, F. et al. Understanding alternative splicing: towards a cellular code. Nature Reviews Molecular Cell Biology 6, 386–398 (2005). doi: 10.1038/nrm1645
Matsuyama, T., Tamaoki, M. et al. CDNA microarray assessment for ozone-stressed Arabidopsis thaliana. Environmental Pollution 117, 191–194 (2002). doi: 10.1016/S0269-7491(01)00320-7
Medini, D., Serruto, D. et al. Microbiology in the post-genomic era. Nature Reviews Microbiology 6, 419–430 (2008). doi: 10.1038/nrmicro1901
Modrek, B. & Lee, C. A genomic view of alternative splicing. Nature Genetics 30, 13–19 (2002).
Morozova, O. & Marra, M. A. Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264 (2008). doi: 10.1016/j.ygeno.2008.07.001
Niculescu, A. B., III & Kelsoe, J. R. Convergent functional genomics: application to bipolar disorder. Annals of Medicine 33, 263–271 (2001).
Noonan, J. P. Neanderthal genomics and the evolution of modern humans. Genome Research 20, 547–553 (2010).
Ouzounis, C. & Mazière, P. Maps, books and other metaphors for systems biology. BioSystems 85, 6–10 (2006). doi: 10.1016/j.biosystems.2006.02.007
Patil, K. R., Åkesson M. et al. Use of genome-scale microbial models for metabolic engineering. Current Opinion in Biotechnology 15, 64–69 (2004). doi: 10.1016/j.copbio.2003.11.003
Pennie, W. D., Woodyatt, N. J. et al. Application of genomics to the definition of the molecular basis for toxicity. Toxicology Letters 120, 353–358 (2001). doi: 10.1016/S0378-4274(01)00322-8
Petsko, G. A. The Rosetta stone. Genome Biology 2, (comment1007) (2001).
Poinar, H. N., Schwarz, C. et al. Metagenomics to Paleogenomics: Large-Scale Sequencing of Mammoth DNA. Science 311, 392–394 (2006). doi: 10.1126/science.1123360
Rabilloud, T. Two-dimensional gel electrophoresis in proteomics: old, old fashioned, but it still climbs up the mountains. Proteomics 2, 3–10 (2002).
Ronaghi, M., Uhlen, M. et al. A sequencing method based on real-time pyrophosphate. Science 281, 363–365 (1998). doi: 10.1126/science.281.5375.363
Rusch, D. B., Halpern, A. L. et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. Public Library of Science Biology 5, e77 (2007). doi: 10.1371/journal.pbio.0050077
Sanger, F., Nicklen, S. et al. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Science, USA 74, 5463–5467 (1977).
Scholin, C. A. What are "ecogenomic sensors?" A review and thoughts for the future. Ocean Science 6, 51–60 (2010). www.ocean-sci.net/6/51/2010/
Schrader, W. P., A metaphor for the map. Science 292, 856 (2001).
Sekirov, I., Russell, S. L. et al. Gut microbiota in health and disease. Physiological Reviews 90, 859–904 (2010). doi: 10.1152/physrev.00045.2009
Singer, E. The $100 genome. Technology Review (2008).
Stoughton, R. B. Applications of DNA microarrays in biology. Annual Review of Biochemistry 74, 53–82 (2005). doi: 10.1146/annurev.biochem.74.082803.133212.
Szostak, J. W., Bartel, D. P. et al. Synthesizing life. Nature 409, 387–390 (2001). doi: 10.1038/35053176
Templin, M. F., Stoll, D. et al. Protein microarrays: Promising tools for proteomic research. Proteomics 3, 2155–2166 (2003). doi: 10.1002/pmic.200300600
Tschöp, M. H., Hugenholtz, P. et al. Getting to the core of the gut microbiome. Nature Biotechnology 27, 344–346 (2009). doi: 10.1038/nbt0409-344
Tyers, M. & Mann, M. From genome to proteome. Nature 422, 193–197 (2003).
Ureta-Vidal, A., Ettwiller, L. et al. Comparative genomics: genome-wide analysis in metazoan eukaryotes. Nature Reviews Genetics 4, 251–262 (2003). doi: 10.1038/nrg1043
Vandepoele, K., Raes, J. et al. Genome-wide analysis of core cell cycle genes in Arabidopsis. The Plant Cell 14, 903–916 (2002).
Venter, J. C., Adams, M. D. et al. The sequence of the human genome. Science 291, 1304–1351 (2001). doi: 10.1126/science.1058040
Venter, J. C., Remington, K. et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74 (2004). doi: 10.1126/science.1093857
Vukmirovic, O. G. & Tilghman, S. M., Exploring genome space. Nature 405, 820–822 (2000). doi: 10.1038/35015690
Wheeler, D. A., Srinivasan, M. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–877 (2008). doi: 10.1038/nature06884
Woese, C. R. & Fox, G. E. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Sciences 74, 5088–5090 (1977).










