


















« Prev Next »
DNA replication is a truly amazing biological phenomenon. Consider the countless number of times that your cells divide to make you who you are—not just during development, but even now, as a fully mature adult. Then consider that every time a human cell divides and its DNA replicates, it has to copy and transmit the exact same sequence of 3 billion nucleotides to its daughter cells. Finally, consider the fact that in life (literally), nothing is perfect. While most DNA replicates with fairly high fidelity, mistakes do happen, with polymerase enzymes sometimes inserting the wrong nucleotide or too many or too few nucleotides into a sequence. Fortunately, most of these mistakes are fixed through various DNA repair processes. Repair enzymes recognize structural imperfections between improperly paired nucleotides, cutting out the wrong ones and putting the right ones in their place. But some replication errors make it past these mechanisms, thus becoming permanent mutations. These altered nucleotide sequences can then be passed down from one cellular generation to the next, and if they occur in cells that give rise to gametes, they can even be transmitted to subsequent organismal generations. Moreover, when the genes for the DNA repair enzymes themselves become mutated, mistakes begin accumulating at a much higher rate. In eukaryotes, such mutations can lead to cancer.
Errors Are a Natural Part of DNA Replication
After James Watson and Francis Crick published their model of the double-helix structure of DNA in 1953, biologists initially speculated that most replication errors were caused by what are called tautomeric shifts. Both the purine and pyrimidine bases in DNA exist in different chemical forms, or tautomers, in which the protons occupy different positions in the molecule (Figure 1). The Watson-Crick model required that the nucleotide bases be in their more common "keto" form (Watson & Crick, 1953). Scientists believed that if and when a nucleotide base shifted into its rarer tautomeric form (the "imino" or "enol" form), a likely result would be base-pair mismatching. But evidence for these types of tautomeric shifts remains sparse.

Figure 1: Tautomeric shifts in nucleotide bases.
The purine and pyrimidine bases in DNA exist in two different tautomers, or chemical forms. (A) Nucleotide bases shift from their common “keto” form to their rarer, tautomeric “enol” form. (B) In common base pair arrangements, the common form of thymine (T) binds with the common form of adenine (A), and the common form of cytosine (C) binds with the common form of guanine (G). (C) Rare base-pairing arrangements result when one nucleotide in a base pair is the rare form instead of the common form. Here, the rare form of cytosine binds to the common form of adenine instead of guanine. The rare form of guanine binds to the common form of thymine instead of cytosine.
© 2014 Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved. 
Today, scientists suspect that most DNA replication errors are caused by mispairings of a different nature: either between different but nontautomeric chemical forms of bases (e.g., bases with an extra proton, which can still bind but often with a mismatched nucleotide, such as an A with a G instead of a T) or between "normal" bases that nonetheless bond inappropriately (e.g., again, an A with a G instead of a T) because of a slight shift in position of the nucleotides in space (Figure 2). This type of mispairing is known as wobble. It occurs because the DNA double helix is flexible and able to accommodate slightly misshaped pairings (Crick, 1966).

Figure 2: Wobble in mismatched nucleotide base pairs.
A shift in the position of nucleotides causes a wobble between a normal thymine and normal guanine. An additional proton on adenine causes a wobble in an adenine-cytosine base-pair.
© Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved.
Replication errors can also involve insertions or deletions of nucleotide bases that occur during a process called strand slippage. Sometimes, a newly synthesized strand loops out a bit, resulting in the addition of an extra nucleotide base (Figure 3). Other times, the template strand loops out a bit, resulting in the omission, or deletion, of a nucleotide base in the newly synthesized, or primer, strand. Regions of DNA containing many copies of small repeated sequences are particularly prone to this type of error.
Fixing Mistakes in DNA Replication
DNA polymerase enzymes are amazingly particular with respect to their choice of nucleotides during DNA synthesis, ensuring that the bases added to a growing strand are correctly paired with their complements on the template strand (i.e., A's with T's, and C's with G's). Nonetheless, these enzymes do make mistakes at a rate of about 1 per every 100,000 nucleotides. That might not seem like much, until you consider how much DNA a cell has. In humans, with our 6 billion base pairs in each diploid cell, that would amount to about 120,000 mistakes every time a cell divides!
Fortunately, cells have evolved highly sophisticated means of fixing most, but not all, of those mistakes. Some of the mistakes are corrected immediately during replication through a process known as proofreading, and some are corrected after replication in a process called mismatch repair. When an incorrect nucleotide is added to the growing strand, replication is stalled by the fact that the nucleotide's exposed 3′-OH group is in the "wrong" position. (Recall that new nucleotides are added to the growing strand during replication by means of their 5′-phosphate group binding to the 3′-OH group of the previous nucleotide on the strand.) During proofreading, DNA polymerase enzymes recognize this and replace the incorrectly inserted nucleotide so that replication can continue. Proofreading fixes about 99% of these types of errors, but that's still not good enough for normal cell functioning.
After replication, mismatch repair reduces the final error rate even further. Incorrectly paired nucleotides cause deformities in the secondary structure of the final DNA molecule. During mismatch repair, enzymes recognize and fix these deformities by removing the incorrectly paired nucleotide and replacing it with the correct nucleotide.
When Replication Errors Become Mutations
Incorrectly paired nucleotides that still remain following mismatch repair become permanent mutations after the next cell division. This is because once such mistakes are established, the cell no longer recognizes them as errors. Consider the case of wobble-induced replication errors. When these mistakes are not corrected, the incorrectly sequenced DNA strand serves as a template for future replication events, causing all the base-pairings thereafter to be wrong. For instance, in the lower half of Figure 2, the original strand had a C-G pair; then, during replication, cytosine (C) is incorrectly matched to adenine (A) because of wobble. In this example, wobble occurs because A has an extra hydrogen atom. In the next round of cell division, the double strand with the C-A pairing would separate during replication, each strand serving as a template for synthesis of a new DNA molecule. At that particular spot, C would pair with G, forming a double helix with the same sequence as its original (i.e., before the wobble occurred), but A would pair with T, forming a new DNA molecule with an A-T pair in place of the original C-G pair. This type of mutation is known as a base, or base-pair, substitution. Base substitutions involving replacement of one purine for another or one pyrimidine for another (e.g., a mismatched A-A pair, instead of A-T) are known as transitions; the replacement of a purine by a pyrimidine, or vice versa, is called a transversion.
Likewise, when strand-slippage replication errors are not corrected, they become insertion and deletion mutations. Much of the early research on strand-slippage mutations was conducted by George Streisinger in the 1970s. Streisinger, a professor at the University of Oregon and a fish hobbyist, is known by some as the "founding father of zebrafish research." However, he is also known for his work with phage T4, a bacterial virus. Streisinger used this virus to show that most nucleotide insertion and deletion mutations occur in areas of DNA that contain many repeated sequences (also called tandem repeats), and he formulated the strand-slippage hypothesis to explain why this was the case (Streisinger et al., 1966). (In Figure 3, notice the series of repeat T's on the template strand where the slippage has occurred.) When slippage takes place, the presence of nearby duplicate bases stabilizes the slippage so that replication can proceed. During the next round of replication, when the two strands separate, the insertion or deletion on either the template or primer strand, respectively, will be perpetuated as a permanent mutation. Scientists have collected enough evidence to confirm Streisinger's strand-slippage hypothesis, and this type of mutagenesis remains an active field of scientific research.
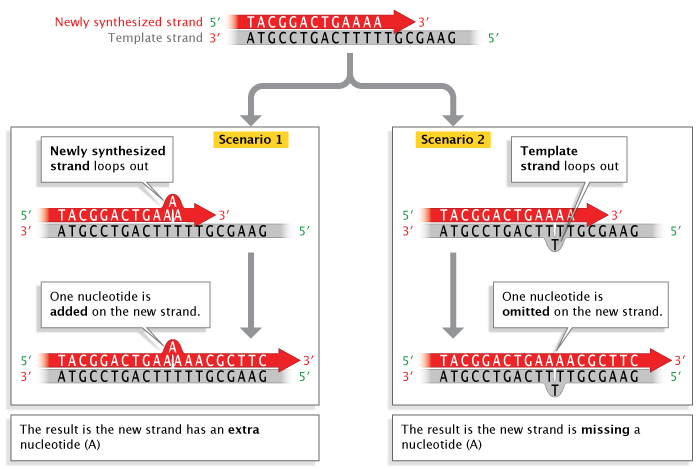
Figure 3: Strand slippage during DNA replication.
When strand slippage occurs during DNA replication, a DNA strand may loop out, resulting in the addition or deletion of a nucleotide on the newly-synthesized strand.
© 2014 Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved.
Although most mutations are believed to be caused by replication errors, they can also be caused by various environmentally induced and spontaneous changes to DNA that occur prior to replication but are perpetuated in the same way as unfixed replication errors. As with replication errors, most environmentally induced DNA damage is repaired, resulting in fewer than 1 out of every 1,000 chemically induced lesions actually becoming permanent mutations. The same is true of so-called spontaneous mutations. "Spontaneous" refers to the fact that the changes occur in the absence of chemical, radiation, or other environmental damage. Rather, they are usually caused by normal chemical reactions that go on in cells, such as hydrolysis. These types of errors include depurination, which occurs when the bond connecting a purine to its deoxyribose sugar is broken by a molecule of water, resulting in a purine-free nucleotide that can't act as a template during DNA replication, and deamination, which results in the loss of an amino group from a nucleotide, again by reaction with water. Again, most of these spontaneous errors are corrected by DNA repair processes. But if this does not occur, a nucleotide that is added to the newly synthesized strand can become a permanent mutation.
Even Low Mutation Rates Can Be Cause for Concern
Mutation rates vary substantially among taxa, and even among different parts of the genome in a single organism. Scientists have reported mutation rates as low as 1 mistake per 100 million (10-8) to 1 billion (10-9) nucleotides, mostly in bacteria, and as high as 1 mistake per 100 (10-2) to 1,000 (10-3) nucleotides, the latter in a group of error-prone polymerase genes in humans (Johnson et al., 2000).
Even mutation rates as low as 10-10 can accumulate quickly over time, particularly in rapidly reproducing organisms like bacteria. This is one reason why antibiotic resistance is such an important public health problem; after all, mutations that accumulate in a population of bacteria provide ample genetic variation with which to adapt (or respond) to the natural selection pressures imposed by antibacterial drugs (Smolinski et al., 2003). Take E. coli, for example. The genome of this common intestinal bacterium has about 4.2 million base pairs, or 8.4 million bases. Assuming a mutation rate of 10-9 (i.e., midway between reported estimates of 10-8 and 10-10), every time E. coli divides, each daughter cell will have, on average, 0.0084 new mutations. Or, another way to think about it is like this: Approximately 1% of bacterial cells will contain a new mutation. That may not seem like much. However, because bacteria can divide as rapidly as twice per hour, a single bacterium can grow into a colony of 1 million cells in only about 10 hours (220 = 1,048,576). At that point, approximately 10,000 of these bacteria will have accumulated at least one mutation. As the number of bacteria carrying different mutations increases, so too does the likelihood that at least one of them will develop a drug-resistant phenotype.
Likewise, in eukaryotes, cells accumulate mutations as they divide. In humans, if enough somatic mutations (i.e., mutations in body cells rather than sperm or egg cells) accumulate over the course of a person's lifetime, the end result could be cancer. Or, less frequently, some cancer mutations are inherited from one or both parents; these are often referred to as germ-line mutations. One of the first cancer-associated somatic mutations was discovered in 1982, when researchers found that a mutated HRAS gene was associated with bladder cancer (Reddy et al., 1982). HRAS encodes for a protein that helps regulate cell division. Since then, scientists have identified several hundred additional "cancer genes." Some of them, like the handful of germ-line mutations associated with a form of colorectal cancer known as hereditary nonpolyposis colorectal cancer (HNPCC), play crucial roles in DNA repair (Wijnen et al., 1998).
Of course, not all mutations are "bad." But, because so many mutations can cause cancer, DNA repair is obviously a crucially important property of eukaryotic cells. However, too much of a good thing can be dangerous. If DNA repair were perfect and no mutations ever accumulated, there would be no genetic variation—and this variation serves as the raw material for evolution. Successful organisms have thus evolved the means to repair their DNA efficiently but not too efficiently, leaving just enough genetic variability for evolution to continue.
References and Recommended Reading
Crick, F. H. S. Codon-anticodon pairing: The wobble hypothesis. Journal of Molecular Biology 19, 548–555 (1966) (link to article)
Johnson, R. E., et al. Fidelity of human DNA polymerase η. Journal of Biological Chemistry 275, 7447–7450 (2000)
Reddy, E. P., et al. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300, 149–152 (1982) (link to article)
Smolinski, M., et al. Microbial Threats to Health: Emergence, Detection, and Response (Washington, D.C., National Academies Press, 2003)
Streisinger, G., et al. Frameshift mutations and the genetic code. Cold Spring Harbor Symposia on Quantitative Biology 31, 77–84 (1966)
Watson, J. D., & Crick, F. H. S. Molecular structure of nucleic acids. Nature 171, 737–738 (1953) (link to article)
Wijnen, J., et al. MSH2 genomic deletions are a frequent cause of HNPCC. Nature Genetics 20, 326–328 (1998) (link to article)










