


 Table 1: Public plant genome sequencing projects.
Table 1: Public plant genome sequencing projects.
















« Prev Next »
Early efforts at sequencing the genomes of bacteria, viruses, and yeast allowed scientists to test and troubleshoot methods of automated sequencing, chromosome assembly, and gene annotation. Using these methods, researchers were able to sequence several unicellular genomes by the year 2000. However, many interesting biological questions remained, and a great deal of these questions related to issues involving development, disease, and complex traits that are found only in multicellular organisms. For example, how are limbs and tissues encoded in the genome? How is the brain made? What are the genetic contributions to behavior? Today, researchers are deeply involved in the shotgun sequencing of various complex genomes in an attempt to answer such questions. Interestingly, they have found that neither genome size nor number of genes determines the complexity of multicellular organisms.
Whole-Genome Shotgun Sequencing
Unicellular genomes generally lack repetitive regions that are difficult to sequence, and these genomes are easily assembled into chromosomes. Multicellular genomes, by contrast, are more difficult to clone, sequence, and assemble. Because of the expense and slow pace associated with clone-based sequencing, researchers have mainly relied on the "shotgun" method of sequencing for multicellular genomes. The whole-genome shotgun (WGS) method entails sequencing many overlapping DNA fragments in parallel and then using a computer to assemble the small fragments into larger contigs and, eventually, chromosomes (Figure 1). This method has the advantage of simplicity and rapidity and works best for genomes with fewer repeated regions. Genomes containing lots of repetitive sequences (like the human genome) create difficulties with chromosome assembly because the computer cannot tell which unique location to map identical DNA sequences to. The hybrid WGS method overcomes this problem by breaking the genome into overlapping clones that can also be physically mapped to the genome, and then performing shotgun sequencing on these intermediate segments. The result is a large-scale map that tells the exact order for each piece of sequenced DNA.
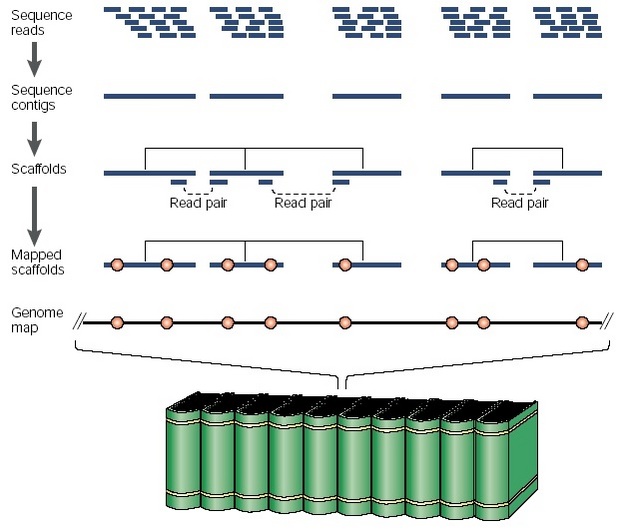
Figure 1: Long-range sequence assembly in whole-genome shotgun sequencing.
Individual sequence reads generated in a whole-genome shotgun-sequencing project are initially assembled into sequence contigs. Groups of sequence contigs are then organized into scaffolds on the basis of linking information provided by read pairs (in each case, with one sequence read from a pair assembling into one contig and the other read into another contig). In turn, the scaffolds can be aligned relative to the source genome (represented by an encyclopedia set) by the identification of already mapped, sequence-based landmarks (for example, STSs, genetic markers and genes; depicted as red circles) in the sequence contigs, thereby associating them with a known location on the genome map.
© 2001 Nature Publishing Group Green, E. D. Strategies for the systematic sequencing of complex genomes. Nature Reviews Genetics 2, 580 (2001). All rights reserved. 
Roundworm and Fruit Fly Genome Sequences
The first sequenced metazoan genomes—those of the fruit fly and the roundworm—were instrumental in the development of the complex assembly and annotation software required to analyze large genomes. Thus, these model organisms were the testing ground for the new sequencing and analysis technologies that would be required to complete the Human Genome Project on time.
The genome of the nematode roundworm C. elegans was sequenced in 1998 by a publicly funded collaborative team based primarily at two sites: Washington University in the United States, and the Sanger Center in the United Kingdom. The research team eventually determined that this simple organism had 18,000 genes, at least a thousand of which were different olfactory receptors (C. elegans Sequencing Consortium, 1998).
Next, the genome of the fruit fly D. melanogaster was sequenced in 2000 by collaboration between the private company Celera and the public Berkeley Drosophila Genome Project (BDGP) based in California (Adams et al., 2000). Surprisingly, the more complex fly had fewer genes than C. elegans—only 13,600. The size of some gene families also varied greatly. For example, while the roundworm had 1,000 genes for smell, the fly had only 60, providing a clue, perhaps, to the relative importance of this sensory pathway in the two organisms (Rubin et al., 2000).
Perhaps most interestingly, the BDGP and Celera researchers determined that about one-third of the genes in Drosophila undergo alternative splicing and thus end up coding multiple proteins. The fly, therefore, can make more than 20,000 proteins with less than 14,000 genes. Worms also perform alternative splicing, but only 13% of worm genes have alternate splice forms identified. Alternative splicing is a mechanism in humans as well, and it may explain how a surprisingly small number of human genes can give rise to the great complexity of the human body. Indeed, scientists estimate that 40%–60% of human genes are alternatively spliced. Furthermore, in-depth analysis of Drosophila genes has revealed that at least 60% of known human disease and cancer genes have related sequences in the fly (Rubin et al., 2000). The Alzheimer's gene is just one example of numerous human disease genes that have been studied extensively in fruit flies.
In addition to learning how many genes are shared among flies, worms, and mammals, the BDGP also evaluated the number and types of distinct protein families that each model organism contains. They discovered that those proteins found in worms and flies that are not present in yeast are associated with multicellular developmental processes, such as cell adhesion and cell-to-cell signaling. Among the large protein families that are present only in flies are proteins involved in the immune response and those that are probably fly-specific, such as cuticle proteins and larval serum proteins (Rubin et al., 2000).
Rice and Mice
Following Drosophila and C. elegans, Arabidopsis thaliana was the first plant and the third multicellular organism to be completely sequenced (Arabidopsis Genome Initiative, 2000). The goals of Arabidopsis sequencing included informing research on crop plants and delineating evolutionary relationships among plant families. However, many researchers have pointed out that Arabidopsis is not closely related to many plants of economic interest. Thus, the recent completion of a high-quality rice genome sequence provides an opportunity to apply research results more directly to crop plants (International Rice Genome Sequence Project, 2005). Comparison of the gene products of rice and Arabidopsis shows that 71% of rice proteins are reasonably similar to Arabidopsis proteins (Table 1; Bevan & Walsh, 2005). This promising and unexpectedly high similarity suggests that Arabidopsis may indeed be a model organism for at least one crop plant.
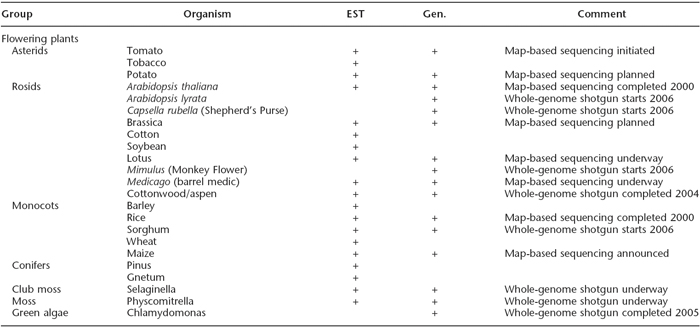
© 2005 Cold Spring Harbor Laboratory Press Bevan, M. et. al., The Arabidopsis genome: A foundation for plant research, Genome Research 15, 1632-1642 (2005). All rights reserved.
Similarity between species was also a critical finding in studies involving the mouse genome. This genome was sequenced in parallel with the human genome and completed in 2002 by the Mouse Genome Sequencing Consortium, which was based at both the Massachusetts Institute of Technology and Washington University in St. Louis. Comparison between human and mouse genomes revealed several interesting points. At the nucleotide level, approximately 40% of the human genome can be aligned to the mouse genome. But when the genomes are partitioned into corresponding regions of conserved synteny, the similarity increases to 90%. These segments likely tell the story of the species' most recent shared ancestor.
The Mouse Genome Sequencing Consortium (2002) also discovered that the mouse and human genomes each contain about 30,000 protein-coding genes (Figure 2). Moreover, the proportion of mouse genes with a single clear homologue in the human genome was estimated to be approximately 80%. Nonetheless, the researchers did identify some mouse-specific characteristics of the animal's genome, describing these findings as follows:
"Dozens of local gene family expansions have occurred in the mouse lineage. Most of these seem to involve genes related to reproduction, immunity, and olfaction, suggesting that these physiological systems have been the focus of extensive lineage-specific innovation in rodents."
Platypus Puzzle
When the genome for the platypus was sequenced (Warren et al., 2008), comparative genomics was put to its strangest test yet, because the platypus is a very strange animal. Classified as a mammal because it makes milk and has fur, the platypus also possesses features of reptiles and birds, such as egg laying. Furthermore, the animal's mouth physically resembles a duck's bill, and males can deliver snake-like venom through spurs on their legs.
In platypus DNA, scientists found genes for egg laying—a feature of reptiles—as well as for lactation—a characteristic of all mammals. The researchers also noted that genetic sequences responsible for venom production in the male platypus appear to have arisen from duplications in a group of genes that evolved from ancestral reptile genomes. Further study of this odd puzzle piece of a genome will help scientists see the big picture of mammalian evolution from a novel perspective.
Lessons Learned
Thus, the number of genes in many simple multicellular model organisms has turned out to be not that different than the number in humans (Figure 3). The lesson to be learned from this information is that genome size does not predict the number of genes, and that neither the size of the genome nor the number of genes can be correlated with the complexity of an organism. Multicellular genomes have yet to reveal all of their secrets, however. While comparison to unicellular genomes has helped define the basic genes required for all life, the genes and splice variants found in complex metazoan genomes promise to elucidate how limbs, organs, and behaviors are sculpted. As the genomes of more and more species are sequenced, scientists will gain even greater insight as to how to read the genomic blueprint and understand how life is encoded in DNA.
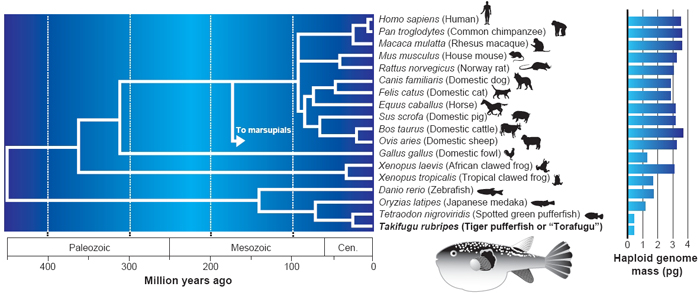
© 2002 Sudhir Kumar and S. B. Hedges. Used with permission. All rights reserved.
References and Recommended Reading
Adams, M. D., et al. The genome sequence of Drosophila melanogaster. Science 287, 2185–2195 (2000) doi:10.1126/science.287.5461.2185.
Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000) (link to article)
Bevan, M., & Walsh, S. The Arabidopsis genome: A foundation for plant research. Genome Research 15, 1632–1642 (2005).
C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282, 2012–2018 (1998) doi:10.1126/science.282.5396.2012.
Guénet, J. L. The mouse genome. Genome Research 15, 1729–1740 (2005).
Green, E. D. Strategies for the systematic sequencing of complex genomes. Nature Reviews Genetics 2, 573–583 (2001) doi:10.1038/35084503 (link to article)
Hedges, S. B. & Kumar, S. Vertebrate genomes compared. Science 297, 1283-1285 (2002).
International Rice Genome Sequence Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002) (link to article)
Rubin, G. M., et al. Comparative genomics of the eukaryotes. Science 287, 2204–2215 (2000) doi:10.1126/science.287.5461.2204.
Warren, W. C., et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature 453, 175–183 (2008) doi:10.1038/nature06936 (link to article)










