


















« Prev Next »
Cancer rates increase markedly with age. This is a major clue that there is some connection between aging and the regulation of cell division. While childhood cancers are rare, cancers among the elderly are common. The few treatments that have been found to slow aging, such as caloric restriction and dampened insulin/insulin-like growth factor 1 signaling, also delay the onset of cancer. This common outcome is somewhat unexpected, as cancer is marked primarily by uncontrolled cell division, an immortal cellular state.
The Dawn of Cellular Aging Research
In 1961, cellular aging was first described by Hayflick and Moorhead. They showed that human cells in culture do not divide indefinitely but reach a limit (called the Hayflick limit) of replication and stop all further division. Cells approach this limit by slowing their divisions and entering cellular senescence, a dormant period. Recently, for damaged cells, this pathway of cellular progression has been considered an alternative to apoptosis (cell suicide). Both DNA damage and insufficient telomere replication are common signals leading to these events. When the cell does not trigger either of these pathways, it can become cancerous (Campisi & Yaswen 2009; Campisi 2009) (Figure 1).
Signals leading to formation of tumors can initiate two different pathways. The first is a pathway involving the p53 tumor suppressor protein, coded by the p53 gene, and the second includes another tumor suppressor, the retinoblastoma protein (Rb), coded by the retinoblastoma gene (Rb). Both pathways lead to a transient arrest in cell division and then to apoptosis or to senescence. If an error in signaling causes the cell not to undergo apoptosis or to enter senescence, then these pathways also result in tumor formation.
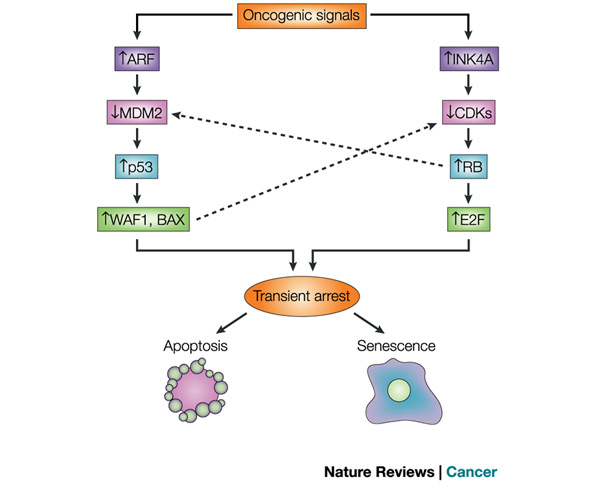
Figure 1: The p53 and RB tumour-suppressor pathways
p53 and RB are at the heart of the two main tumour-suppressor pathways that control cellular responses to potentially oncogenic stimuli. Each pathway consists of several upstream regulators and downstream effectors. For simplicity, only four main components in each pathway are shown. Similarly, the pathways interact at several points, two of which are shown. In the p53 pathway, signals such as DNA damage induce the ARF (also known as p14 in humans and p19 in mice) product of the CDKN2A locus. ARF increases p53 levels by sequestering MDM2, which facilitates the degradation and inactivation of p53. p53 has both transactivation and transrepression activity, and so controls the transcription of numerous genes. Among the p53 target genes are WAF1, an inhibitor of cyclin-dependent protein kinases (CDKs) that, among other activities, causes cell-cycle arrest, and BAX, which promotes apoptotic cell death. In the RB pathway, stress signals such as oncogenes induce INK4A, the other product of the CDKN2A locus. INK4A inhibits CDKs that phosphorylate, and therefore inactivate, RB during the G1 phase of the cell cycle. RB also controls the expression of numerous genes, although it does so primarily by recruiting transcription factors and chromatin remodelling proteins. One downstream consequence of RB activity is the inhibition of E2F activity, which is important for the transcription of several genes that are required for progression through the G1 and S phases of the cell cycle. RB also regulates p53 activity through a trimeric p53-MDM2-RB complex
© 2003 Nature Publishing Group Campisis, J. Cancer and ageing: rival demons? Nature Reviews Cancer 3, 339–349 (2003) doi:10.1038/nrc1073. All rights reserved. 
How Do We Know That Tumor Suppressors Affect Aging?
An important part of understanding how these systems work to control cell division was the discovery that p53 impacts both cancer and aging. Tyner et al. (2002) devised a genetic strategy in mice to compare the effects of the absence of p53 or a smaller than normal p53 protein. The two mutant transgenic mouse lines had either a complete deletion of the p53 gene (p53-) or a truncated form of p53 (p53m, mutant) that did not have the first six exons of the p53 gene (Figure 2).
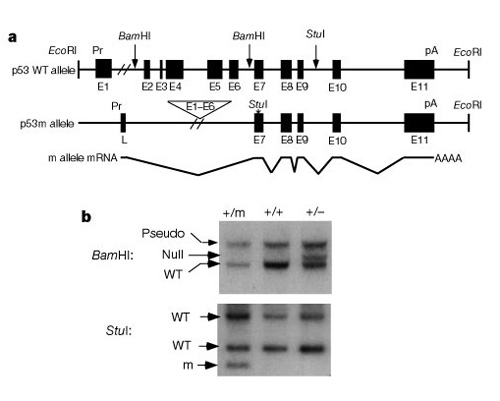
Figure 2: Structure and expression of the mutant p53 m allele
(A) Maps of the p53 wild-type (WT) and m alleles: Exons 1–6 and upstream sequences are deleted in the p53 m allele, juxtaposing exons 7–11 to an unknown promoter that initiates a chimaeric mRNA containing a 55-nucleotide leader (L) spliced into exons 7–11. Pr, promoter; pA, polyadenylation site. (B) Southern analysis of tail DNAs from p53+/+, p53+/- and p53+/m mice: The top panel shows tail DNAs cleaved with BamHI and hybridized to a p53 exon 2–6 probe to differentiate p53 + and - alleles.
© 2002 Nature Publishing Group Tyner, S. D. et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53 (2002) doi:10.1038/415045a. All rights reserved.
The first half of the study compared three groups of mice: group 1, p53+/p53-(deletion of one copy of p53); group 2, p53+/p53m (partial deletion mutant); and group 3, p53+/p53+ (wild type, normal). Table 1 shows these three groups and their experimental results related to the cancer and aging phenotypes. Interestingly none of the mice in group 2, with the truncated p53 protein, developed life-threatening tumors, while 45% of group 3 (wild type) and over 80% of group 1 developed life-threatening tumors. Group 2 mice also had an intermediate life span midway between the very short life span of group 1 and the longer life span of group 3 wild type mice. The conclusion from these data is that the partial p53 mutation reduced the incidence of cancer, and at the same time appeared to cause a deficit in life span, not an extended life span.
| Table 1. Experimental results from genetic mouse studies with p53 mutants | |||
| Genotype | Cancer phenotype | Aging phenotype | |
| Group 1 | p53+/p53- (complete deletion) | 80% had tumors | Much shorter life span |
| Group 2 | p53+/p53m (partial mutant) | None | Shorter life span |
| Group 3 | p53+/p53+ (wild type) | 45% had tumors | Normal life span |
|
Adapted from Tyner et
al. 2002 | |||
The authors also noted that the group 2 mice developed phenotypes characteristic of old mice, such as slow hair regrowth and hunchbacked spines due to skeletal changes, earlier than the wild-type mice did (Figure 3).

Figure 3: Survival trends for mice with various forms of p53 gene
More than half of the tumours in wild-type mice appeared while many of the p53+/m mice were still alive (a). Virtually all of the p53-/- and p53-/m mice developed cancer (b).
© 2002 Nature Publishing Group Tyner, S. D. et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53 (2002) doi:10.1038/415045a. All rights reserved.
In the second half of the study, Tyner and colleagues asked whether the p53 mutant functioned differently in the presence of normal p53. They bred an additional transgenic mouse line with p53-/p53m mutant alleles and found that these mice did not have the strong tumor protection and displayed a much lesser life span effect. So the mutant p53 somehow needed to work in concert with normal p53 to have an effect. As a general observation, the researchers reported that cells from the p53+/ p53m heterozygote turned out to be about three times as hard to transform as wild-type cells. Thus, although these cells were resistant to cancer, this p53+/ p53m background also caused earlier aging. Indeed, the activity of p53 in these heterozygotes appeared to be notably higher than its activity in the wild type. It seemed that such a change, while a priori, would be good against both cancer and senescence, but that turned out not to be the case.
Later, Moore et al. demonstrated that in cultured cells with the same mutation causing truncated p53 protein, this truncated protein entered the nucleus and colocalized with the normal p53. They also studied the p53 protein's half-life within cells and found that the heterozygotes with one copy of the mutant p53 had about a threefold increase in stability of the normal p53 protein, compared to the stability in the wild type alone, meaning stability of the protein was improved over normal. These results in cultured cells extended and confirmed the study conducted by Tyner et al. in mice. Furthermore, this was the first cell division response pathway discovered to work via p53 and clearly modulate both cancer incidence and aging.
In the Rb pathway, which can signal exiting from the cell division cycle, events such as DNA damage or insufficient replication leading to short telomeres at chromosome ends causes decreased CDK signaling. This, in turn, increases Rb protein kinase activity and consequently increases the activity of the transcription factor E2F. What is the significance of E2F? This transcription factor binds to the promoters of RNA polymerase subunits and other proteins needed for the S phase to begin, and it helps start cell division (Campisi 2003; Weinberg 1995). Thus it seems that both the p53 and the Rb pathways impinge on the same cell cycle control mechanisms.
Summary
Aging mammalian cells can stop dividing and enter senescence if they are damaged or have defective telomeres. Senescence protects against tumor formation, and tumor suppressor genes include some that regulate cell division and lead to senescence. Two pathways - one involving the tumor suppressor gene p53, the other involving the tumor suppressor gene RB - lead to a division arrest followed by either apoptosis (cell suicide) or senescence (the stopping of cell division). Studies of mutant p53 genes have supported the idea that increased tumor protection by p53 can shorten life spans of mice.
References and Recommended Reading
Campisi, J. Cancer and ageing: Rival demons? Nature Reviews Cancer 3, 229–249 (2003)
Campisi, J. & d'Adda do Fagagna, F. Cellular senescence: When bad things happen to good cells. Nature Reviews Molecular Cell Biology 8, 729–740 (2007).
Campisi, J. & Yaswen, P. Aging and cancer biology, 2009. Aging Cell 8, 221–225 (2009).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Experimental Cell Research 25, 585–621 (1961).
Moore, L., et al. Aging-associated truncated form of p53 interacts with wild-type p53 and alters p53 stability, localization, and activity. Mechanisms of Ageing and Development 128, 717–730 (2007).
Tyner, S. D., et al. P53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53 (2002).
Weinberg, R. A. The retinoblastoma protein and cell cycle control. Cell 81, 323-330 (1995).










