




", "true", "Figure 4", "Amplified fragment length polymorphisms (AFLPs)", "", "All rights reserved.", '504', '651', 'http://www.nature.com/scitable');)
















« Prev Next »
Introduction
Ecology is inextricably intertwined with the evolutionary history of organisms. Through the process of descent with modification, organisms are continually passing genetic information from one generation to the next, information that is then recorded in the DNA of their descendents. Molecular biology's ability to access this record to better understand the origins of species and the ecological bases of their existence has become a cornerstone of modern ecological research.
In this article we briefly review the molecular tools and methods available to modern ecologists seeking a deeper understanding of the genetic bases of species formation, diversification, and evolutionary adaptation as they interact with ever-changing, complex environments.
PCR: An Ecologist's Best Friend
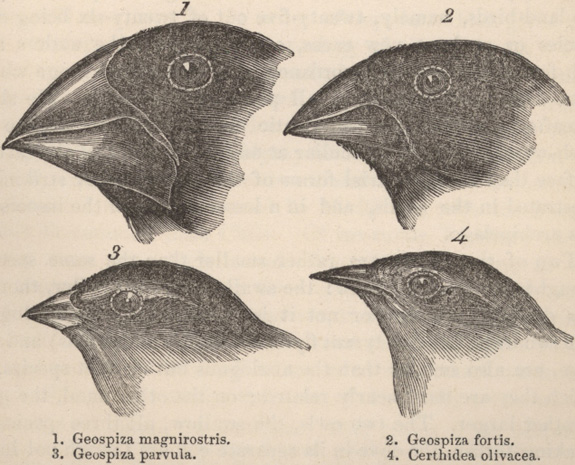
Most molecular-based studies begin with the extraction of DNA from a particular organism, followed by the amplification (i.e., generation of many copies) of particular segments of DNA using the polymerase chain reaction (PCR) (Figure 1). The utility of PCR lies in the fact that only minute quantities of DNA are needed (e.g., nanogram amounts). This is particularly useful when researchers are unable to obtain large amounts of tissue (e.g., as in the case of rare plant or animal species) or when numerous samples are needed, as in the case of population genetic studies. For example, an ecologist might ask: How genetically diverse are populations comprising a single species across a broad environmental gradient? The answer begins by obtaining DNA from different individuals from multiple populations and subjecting it to a PCR-based survey of genetic diversity. Such a survey can lead to inferences about the historical processes that led to differences in the genetic makeup of populations spanning a broad range of geographic and environmental conditions. Alternatively, one might want to know about the evolutionary history and relationships among members of a group of species. Consider, for instance, Darwin's finches. (Figure 2). Once again, PCR is used to amplify particular coding or non-coding regions of DNA from different species, with the ultimate goal of reconstructing the phylogenetic history of each species within the complex. Once determined, the phylogenetic tree resulting from this study can provide information on how diverse the species complex is and which species are most closely related to one another (Figure 3). In turn, this can provide insight into the ecological (e.g., niche space use) and behavioral factors (e.g., foraging) that have contributed to the diversity of a species complex.
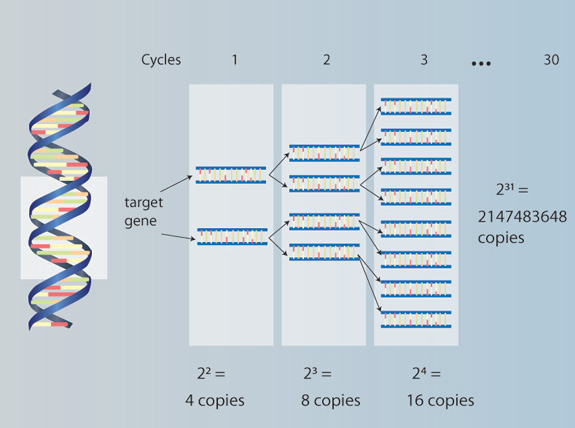
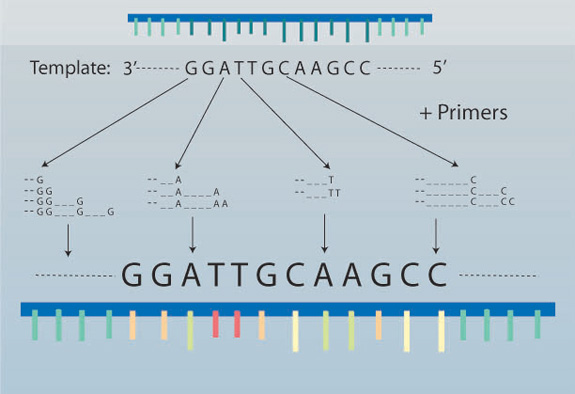
Figure 1: Polymerase chain reaction (PCR)
The PCR method begins with total genomic DNA extracted from an organism. The DNA is combined with site-specific primers, taq polymerase, and other reagents (e.g., MgCl2, buffer, dNTPs) and subjected to repeated cycles, each of which consists of a denaturation phase, annealing phase and extension phase. Denaturation separates double stranded DNA, allowing primers to anneal to specific sites, followed by incorporation of deoxynucleotide triphosphates (dNTPs; A, C, G, T), thereby extending the target site in the 5'-3' direction (on both separated strands). The first cycle is completed when one round of denaturation, annealing and extension is finished, resulting in two new copies of the target site. Subsequent cycles (typically 30-35) repeat the 3-phase process, resulting in many million-fold copies of amplified DNA.
© 2010 Nature Education All rights reserved. 
Markers and Methods
There are many different types of DNA markers used in molecular ecology, including: microsatellites (MSATs, highly repetitive sequences of DNA that mutate rapidly and are often used to identify individuals), minisatellites (similar to microsatellites but with longer repetitive sequences), restriction fragment length polymorphisms (RFLPs, specific sites of DNA that can be cut by enzymes yielding different-sized fragments of DNA in different species, populations, and — rarely — individuals), and DNA sequence data (the bases of DNA are determined and similarities and differences are compared to identify species, populations, and individuals). Markers generated by these methods are also visualized in different ways. Traditionally, MSATs and RFLPs were visualized as discrete bands revealed by agarose gel electrophoresis. The nucleotides comprising DNA sequences, however, require finer levels of resolution, often achieved using polyacrylamide gels and autoradiography. Today, these marker types are typically visualized using chemifluorescence and genetic analyzers, which detect the fluorescent emission of labeled primers (as in the case of MSATs) or the fluorescently-labeled nucleotides of DNA sequences. These markers and visualization methods are by no means a comprehensive list, and the technique one chooses depends greatly on the type of question being addressed in the study. By understanding the different kinds of information provided by different marker methods, one can come to an informed decision on which is best for a particular study. Below, we describe three molecular methods commonly used in molecular ecological studies.
There are three different classes of markers that can be easily distinguished based on the type of information they provide. Anonymous markers include those generated by a method called amplified fragment length polymorphisms (AFLPs) (Figure 4). This technique uses restriction enzymes combined with PCR to generate many thousands of unique fragments that can be used to genetically fingerprint individuals within or among species within the same genus. The utility of the AFLP method lies in that it does not require prior knowledge of an organism's genome. In other words, the regions of the genome that are targeted by this method are unknown to the investigator (hence, "anonymous"). Nevertheless, this method often provides a rich source of information about basic levels of genetic diversity and differentiation. AFLP markers are thus often used as a first step when investigating population or species differences. The downside to the use of AFLPs, however, is that they are somewhat limited in the type of information they can provide. For example, because theses markers are of unknown origin and nucleotide composition (i.e., they simply constitute fragments of varying length within the genome), they are of limited use in reconstructing the evolutionary history of a group of organisms. Furthermore, AFLP markers are commonly referred to as dominant markers and are scored as being either "present" or "absent," which means that it is generally not possible to determine if a band on a gel represents a homozygous (AA) or heterozygous (Aa) genotype . This is because AFLP fragments represent unique restriction sites that are either present or absent in each individual and thus only one allele (if present) is amplfied, thereby limiting the amount of information that can be obtained. Another similar method called random amplified polymorphic DNA (RAPD) also generates dominant markers, which are typically viewed using agarose gel electrophoresis. This method, however, has largely been replaced by the AFLP method, which typically uses chemifluorescence and a genetic analyzer for visualization.
Another class of markers, known as sequence-tagged site (STS) markers, provides an alternative approach to characterizing genetic diversity within and among species. A sequence-tagged site is a short (200-500 bp) sequence of nucleotides that has a unique location within a genome and is targeted using PCR with primers designed by an investigator. One type of STS marker is represented by microsatellites (MSATs), also known as simple sequence repeats (SSRs) or variable number tandem repeats (VNTRs). Unlike AFLPs, these markers do require some knowledge of specific regions containing tandemly repeated nucleotide motifs, such as "ATC" or "GAG," which typically appear in non-coding regions of DNA. In combination with primers specifically designed to target these sites and amplification via PCR, the STS method provides a much finer level of discrimination among individuals. As codominant markers they are able to reveal whether an individual is heterozygous at a particular locus (e.g., Aa v. AA) because both alleles (A and a) are amplified during the PCR process. Given that their exact nucleotide composition (e.g., whether each repeat is always "ATC") is not always known, these markers share the same limitation as AFLPs for phylogenetic reconstruction because the homology of the markers is not known. One way to extend the utility of STS markers whose exact nucleotide composition is unknown is to sequence fragments derived from polymorphic loci. One marker method known as sequence characterized amplified regions (SCARs) uses fragments that have been cloned and sequenced to determine their exact nucleotide composition. Once sequenced, primers can be designed around the SCAR, and then re-amplified to look for fragment length polymorphisms on an agarose gel. Interestingly, this method is often used in combination with anonymous, dominant markers such as AFLPs and RAPDs, thereby also extending their utility.
An alternative, non-PCR-based marker method that is sometimes used by molecular ecologists is allozyme analysis. These markers are derived from loci encoding enzymes used in important metabolic processes (e.g., glycolysis). Although they are not as rapidly evolving as STS markers, they often yield moderate to high levels of genetic variation, depending on the organism. In either case, information from STS or allozyme markers can be used to determine if heterozygosity within populations is correlated with some ecological variable. For example, one could examine levels of heterozygosity relative to growth rate and performance in plants or adaptive response to environmental change in animals.
A third class of markers often used by molecular ecologists are those derived from direct DNA sequencing of targeted regions within the genome. These are often called Sanger sequencing (Figure 5). As with STS markers, DNA sequencing requires precise knowledge of specific genes, or gene regions, that are of interest to the investigator. Combined with PCR and well-designed primers, this method provides the finest and most fundamental level of genetic detail currently available to molecular ecologists. This is because the exact nucleotide sequence can be obtained for cross-comparison analysis of a wide range of taxonomic levels, from phyla to species, and, depending on levels of variation, even among individuals within a population. Thus, DNA sequencing is ideal for determining the evolutionary history of a group of organisms and for inferring evolutionary processes and patterns such as the genetic basis of adaptive trait loci (e.g., genes involved in responses to day length in plants), the historical patterns of migration and expansion of animal species (e.g, from the Pleistocene to present day), and the evolution of specific traits involved in taxonomic diversification (e.g., the origin of a notochord leading to vertebrates) — to name only a few. One particularly useful genome that has been used extensively by molecular ecologists studying animal phylogenetics is the organellar genome of mitochondrial DNA (mtDNA). One region of mtDNA that has proven especially informative at low taxonomic levels (i.e., species level) is the cytochrome oxidase I (COI) region, also known as the "bar-coding" region because of its ability to use universal primers and genetically barcode groups of diverse species. Another genome frequently used by plant molecular ecologists is the chloroplast genome, which has been used extensively to track historical patterns of plant migration and reconstruct plant phylogenies.
DNA sequencing has also enabled the development of another highly polymorphic, codominant marker type called single nucleotide polymorphisms (SNPs). When multiple sequences of a particular region are generated for multiple members within a species, single base differences among individuals are often detected. Depending on the level of DNA sequencing (e.g., individual regions v. whole genomes), SNPs can provide broad genome coverage, show high levels of variability, and can be used for phylogenetic reconstruction because the homology of these markers is known.
Another different but related approach to targeting individual gene regions is whole genome sequencing. One recently developed method that rapidly generates short sequenced segments that can be analyzed and compiled into whole genomes is called Next Generation Sequencing. Although typically limited to organisms with small genomes (e.g., bacteria or viruses), Next Generation Sequencing is becoming an important tool for molecular ecologists interested in probing entire genomes for clues to ecologically-based questions.
Given the strengths and weaknesses of different molecular genetic techniques, one might wonder how best to design an experiment for answering a particular ecological question. This subject requires careful consideration of both marker method and marker information content.
Experimental Design: From Random Molecules to Appropriate Methods
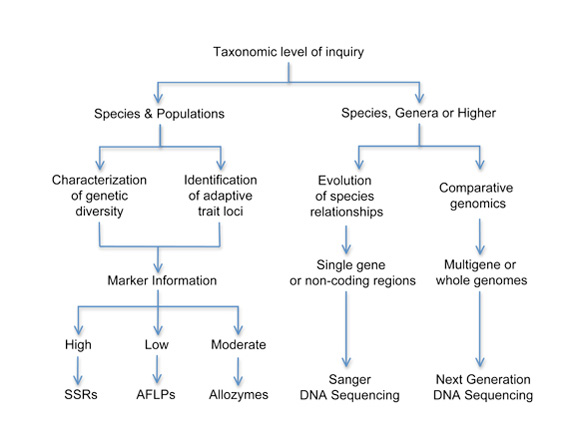
A key question to ask when employing genetic techniques is: Which one is best suited for my particular question? The answer to this question will be determined by several factors, all of which must be evaluated both individually and together in order to arrive at a cohesive plan for launching a successful molecular ecology study. Figure 6 shows a flow diagram for initial consideration of which method (or methods) best apply to your particular question. Although there are many different ways to approach this question, one simple strategy is to begin with the taxonomic level of investigation.
Are you interested in population-level differences within species? Are anonymous markers the only ones available for your organism of interest? If so, then AFLPs might be the marker of choice.
Do you need to know details such as observed or expected heterozygosity? Or, are you trying to correlate neutral marker variation (i.e., ones that are not under selection) with some environmental variable? In this case, AFLPs might be useful, but STS or allozyme markers might be a better way to go.
Are you interested in reconstructing the evolutionary history of a group of organisms? If so, at what level of inquiry is your question aimed: among species within genera, among genera, or at higher taxonomic levels (e.g., families, even phyla)? Depending on the region you intend to target (e.g, coding v. non-coding DNA, nuclear v. organellar mtDNA), homologous markers derived from DNA sequencing will likely provide the greatest dividends.
Do your interests revolve around genome evolution? For example, you might be interested in understanding how the genomes of pathogenic v. non-pathogenic bacteria differ and whether there are ecological or environmental correlates to the virulence of pathogen-related genes.
In this case, whole genome sequencing (not just individual gene regions), which is now feasible and can be easily used to analyze small genomes, would provide a rich source of information for the question of interest.
Although there are multiple ways to assess which marker is best for which question, thinking carefully about what levels of genetic variation you need and at which taxonomic level is paramount to choose the best approach. Understanding this very simple strategy and applying it thoughtfully can ultimately determine the degree to which your question is both answerable and publishable within the field of molecular ecology.










