


















« Prev Next »

Infections with Plasmodium parasites cause human malarias. Among the five species of Plasmodium that infect humans, P. falciparum is the deadliest, causing more than one million malaria-related deaths per year in endemic areas of sub-Saharan Africa, where 90% of all malaria-related deaths occur. Plasmodium parasites are unicellular and eukaryotic organisms. Various species of female Anopheles mosquitoes transmit them. Hence, malaria is a vector-borne disease. All malarial parasites have a complex life cycle that involves sexual and asexual reproductive stages (Figure 1). Parasites reproduce asexually (clonally) in human hosts. The sexual stage takes place inside the mosquito vector. Hence, the genetic material of parasites mixes in the mosquitoes during sexual recombination. The number of sexual reproductions and the amount of sexual recombination increases with the number of mosquitoes that participate in the transmission of the parasite.

Figure 1: Illustration of the malaria transmission cycle.
© 2012 Nature Education All rights reserved. 
History of Drug Resistance
So far, no preventative vaccination against malaria exists, and its control depends heavily upon antimalarial drugs that kill parasites inside the human body. Malaria has been noted for more than 4,000 years. In ancient China, India, the Middle East, Greece, and Rome, malaria and its possible treatments were documented. Ancient Chinese used a treatment based on artemisinin (documented 168 BC), the active ingredient in some high-end drugs nowadays. Treatments with Quinine are known to the western world since the early 17th century. In British colonies, tonic water (which contains large amounts of Quinine) was mixed with gin and became a popular drink.
The discovery of Chloroquine (CQ) in the 1930s revolutionized malaria treatments. CQ was the most widely-used drug from the early 1950s to until the 1990s. After about ten years of use, mutations within P. falciparum that conferred resistance to CQ arose independently in Columbia and Thailand. Since then CQ-resistant mutations have been spreading quickly through most endemic areas. CQ clears out resistant parasites less efficiently from the human body than sensitive (non-resistant) parasites. Hence, infections with resistant parasites result in increased morbidity and mortality.
Sulfadoxine-pyrimethamine (SP), a combination of two drugs, replaced CQ. However, resistance to SP evolved rapidly and now occurs at high frequency in major malarious regions (Laxminarayan 2004). Currently, alternative drugs (e.g., artemisinin-based combination therapies) are available and others continue to be developed. However, higher production costs limit their widespread application in major endemic areas. The evolution of resistance against affordable drugs incurs an enormous societal cost for fighting the spread of the disease. Facing this reality, the focus of public health policy should be shifted to increasing the sustainability of treatment regimes by delaying the emergence and spread of drug resistance as much and early as possible.
Avoiding Drug Resistance
Is it possible to prevent or delay the spread of anti-malaria drug resistance? The answer requires approaches that integrate the disease ecology, epidemiology, genetics, and evolutionary biology of malaria. Such interdisciplinary research started to dissect the dynamics of drug-resistance evolution under various clinical and demographic settings. The complex problem of identifying key determinants in the spread of resistance could be tackled from many starting points. However, a critical path to the heart of the problem is to recognize that the evolution of drug resistance is an example of Darwinian evolution by natural selection.
From the parasites' point of view, drug-treated human hosts represent a novel harsh environment. Any mutation in their genome that reduces the rate at which drugs eliminate them from the host is beneficial to the parasites and will be subject to positive selection. Biological conditions known to delay the propagation of beneficial mutations would provide clues for designing optimal drug-deployment policies that should slow down the spread of resistant parasites. Hence, some theoretical concepts and tools of population genetics to investigate positive selection in plant and animal populations have been employed recently to assist in solving the problem of anti-malarial drug resistance (Escalante et al. 2009).
At first, the population genetic explanation for the emergence of resistance seemed straightforward: in the laboratory, the rate of spontaneous mutation from drug sensitive to resistant alleles was found to be 10-8 per replication. If an infected host carries 1010 parasites in the body, at least 100 of them will be drug-resistant, and their number will keep increasing due to their selective advantage over sensitive parasites within drug-treated hosts. In conclusion, new resistant parasite strains would emerge readily whenever the drug is introduced to an endemic region. However, the analysis of DNA variation among resistant parasites revealed that severe resistance, for both CQ and SP, spread from only a few independent strains worldwide (Roper et al. 2003). This discrepancy arises because the simple evolutionary model above neglects the complexity of the Plasmodium life cycle and the pharmacodynamics of drugs. Furthermore, it points to the problem of conventional population genetics modeling that typically traces the relative frequency, rather than absolute count, of variants over time.
First, despite the abundance of merozoites in the patients' blood, very few of these produce gametocytes that ultimately transfer to mosquitoes (Hastings 2004). Thus the probability that a resistant mutant is included among those gametocytes is very small, unless the relative and absolute frequencies of resistant parasites greatly increase immediately after drug treatment (which is unlikely for several reasons; see below). Second, more importantly, resistance against a particular drug is initially incomplete, but builds up from low to high levels as successive mutations occur within the parasite. An initial resistant mutation creates a strain that survives better than sensitive parasites but is still eliminated under the normal drug concentration (i.e., the initial dose of drug given to the patient). Therefore, the drug kills sensitive and resistant parasites. However, while its absolute number decreases rapidly (say from 100 to 0), resistant parasites may produce a gametocyte that is successfully transferred to a mosquito, which would happen very rarely.
Next, this ‘lucky' resistant parasite in the mosquito still faces elimination: after the mosquito infects a host, a regular drug dosage will likely kill the drug resistant individuals. If the infected host is untreated, drug-sensitive will outcompete drug-resistant parasites (see below). However, the drug concentration in treated hosts decays over time, as does its effect on parasite growth. At an intermediate concentration, resistant parasites can increase their absolute number while the drug kills sensitive parasites. As mosquitoes transfer resistant sporozoites, resistant parasites survive in a host with intermediate drug concentration, and will be further transmitted. If drugs decay slowly, more hosts with intermediate concentration are available, and resistant parasites have more opportunities to establish themselves (Hastings et al. 2002). This may explain why arteminisin, which decays very rapidly, remained effective for more than 25 years in South East Asia.
Once a drug-resistance mutation appears and overcomes the hurdles in initial establishment, its frequency increases rapidly because of its selective advantage over drug-sensitive parasites. This selective advantage depends on the proportion of drug-treated infections, because resistant mutations are advantageous only in drug-treated hosts. When they co-infect an untreated host, drug-sensitive parasites are likely to have an advantage over resistant ones because drug resistance comes with "metabolic costs" that slow down their growth.
These population genetic considerations suggest conditions that would maximally delay the evolution of drug resistance: an anti-malarial drug should be strong enough to kill both sensitive and partially-resistant parasites very quickly. Moreover, the drug should decay fast enough to shorten the time-window of sub-optimal drug concentration. Additionally, comprehensive preventative treatments and the use of counterfeits drugs should be avoided to reduce the selective advantage of resistant mutations (Mackinnon 2005). Furthermore, limited contact between infected hosts and mosquitoes while the drug decays should delay the spread of resistant parasites.
Genetic Signatures of Drug Resistance Evolution
More insight for developing and maintaining an effective drug-deployment policy will come from advanced evolutionary genetic modeling. Policy components will require that the models incorporate key parameters that may determine how fast the resistance spreads. Important factors are geographically-specific variables (e.g., the transmission/migration rate and host immunity) and the evolutionary genetic structure of resistance (e.g., the number and fitness effects of mutated genes and their interactions). Currently only limited information regarding these parameters is available, which severely limits efforts to analyze and evaluate models. One promising approach to obtain the necessary information is to reconstruct the actual events of drug-resistance evolution that recently occurred in various endemic areas. Then scientists can investigate which geographically-specific and genetic variables are associated with the rapid spread of drug resistance. Unfortunately, public health records and samples collected in the past are not comprehensive enough to allow such investigation. For example, year-to-year increase in drug treatment failures in an endemic area - even if accurately documented - cannot be used to calculate the selective advantage of resistance if the proportion of drug-treated infections and other information are not available. However, researchers have indirectly examined the past evolutionary dynamics of drug resistance by applying modern theories of population genetics to present-day samples of malarial parasite DNA. In particular, selective sweeps play a crucial role in inferring basic parameters of drug-resistance evolution.
A selective sweep is the sudden removal of DNA sequence variation at the genomic location of an advantageous gene under strong positive selection (Maynard Smith & Haigh 1974) (Figure 2). By scanning the genome, the discovery of regions of unexpectedly low variation, or another signature of selective sweeps, identified numerous genes that underwent recent adaptive evolution (Nielsen 2005). Notably, some of the most striking examples of selective sweeps are with genes involved in the evolution of anti-malarial drug resistance (Figure 3). Mutations in the pfcrt, dhfr, and dhps genes cause resistance to CQ, pyrimethamine, and sulfadoxine. Scientists observed clear signatures of selective sweeps in parasite samples carrying amino-acid changing mutations in these genes (Wooton et al. 2002, Nair et al. 2003, Nash et al. 2005, Vinayak et al. 2010), as expected from the selective advantage of drug-resistant over drug-sensitive alleles. Furthermore, the exact pattern of selective sweeps might contain information regarding the strength of selection and the past trajectory of the resistant parasites' frequencies. Therefore, by examining selective sweeps in many endemic areas with different demographic and epidemiologic characteristics, we should be able to identify factors determining the relative speed of drug-resistance evolution. Researchers are currently developing evolutionary models that connect major epidemiological variables to the pattern of selective sweeps (Escalante et al. 2009, Schneider & Kim 2010, 2011). These variables include transmission intensities, treatment rates, drug decay rates, immunity (as it relates to drug use), and metabolic costs of resistance.
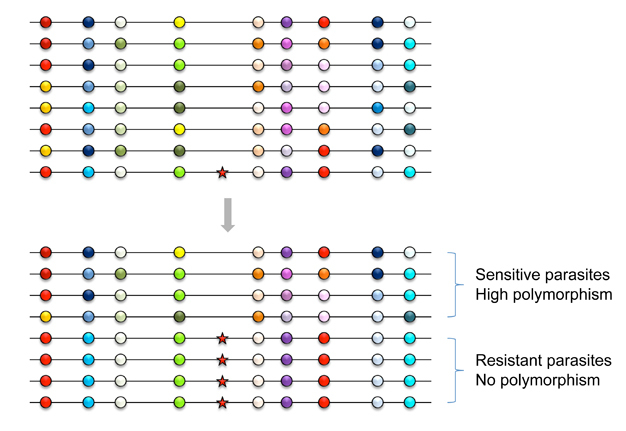
Figure 2: Selective sweep (or hitchhiking effect) of a resistant mutation.
Horizontal lines represent small segments of chromosomes from different parasite individuals in the entire endemic area. Circles denote neutral alleles at polymorphic loci, such as microsatellites. Variation at each locus is depicted by different colors of circles. Before the appearance of the resistant mutation (shown as a red star), the genetic diversity of parasites (high level of polymorphism) is maintained by a long-term balance between new mutations and genetic drift. However, when the drug-resistance mutation increases very rapidly to a high frequency, not only the mutation but also neutral alleles on the same chromosome increase together unless meiotic recombination breaks their association. As a result, no genetic variation is observed around the resistant mutation, causing dichotomy in the level of polymorphism between drug-sensitive and drug-resistant parasites.
© 2012 Nature Education All rights reserved.
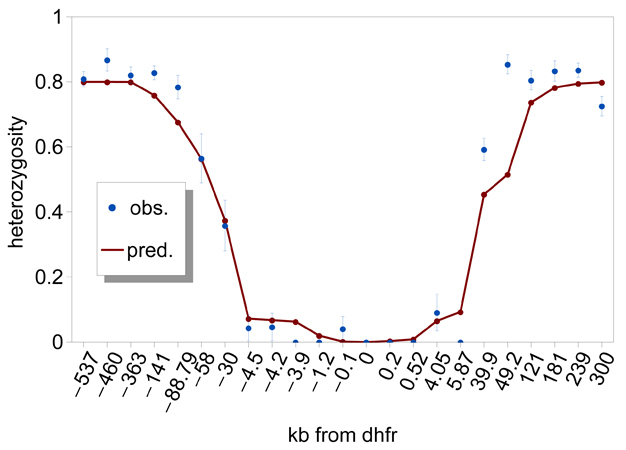
Figure 3: Patterns of genetic variation around the Plasmodium falciparum dhfr gene.
Observed at microsatellite loci that are polymorphic with variable lengths of DNA base repeats, genetic variation is reduced for approximately 100 kb around the dhfr locus on chromosome 4 on the Thailand-Myanmar border (data taken from Nair et al. 2003). The expected heterozygosity, He (±1 s.d.), which is the probability that two sampled chromosomes will carry different alleles at a locus, is plotted against position (distance (kb) of genotyped microsatellite markers relative to dhfr). The solid line shows levels of He predicted by a deterministic hitchhiking model (Schneider & Kim 2010) using parameters that yield a fit to the observation.
© 2012 Nature Education All rights reserved.
In conclusion, applying advanced empirical and theoretical research in population genetics can contribute to the design of a strategy to delay the disaster caused by drug-resistance evolution against newly introduced anti-malarial drugs. These approaches should also be applicable to understanding and reducing the impacts of other infectious diseases.
References and Recommended Reading
Escalante, A. A. et al. The dynamics of mutations associated with anti-malaria drug resistance in Plasmodium falciparum. Trends in Parasitology 25, 557 -563 (2009).
Hastings, I. M. The origins of antimalarial drug resistance. Trends in Parasitology 20, 512 -518 (2004).
Hastings, I. M. et al. The evolution of drug-resistant malaria: The role of drug elimination half-life. Philosophical Transactions of the Royal Society, Series B 357, 505-519 (2002).
Laxminarayan, R. Act now or later? Economics of malaria resistance. American Journal of Tropical Medicine and Hygiene 71, 187-195 (2004).
Mackinnon, M. J. Drug resistance models for malaria. Acta Tropica 94, 207-217 (2005).
Maynard Smith, J. & Haigh, J. The hitch-hiking effect of a favourable gene. Genetics Research 23, 23-35 (1974).
Nair, S. et al. A selective sweep driven by primethamine treatment in Southeast Asian malaria parasites. Molecular Biology and Evolution 20, 1526-1536 (2003).
Nash, D. et al. Selection strength and hitchhiking around two anti-malarial resistance genes. Proceedings of the Royal Society B: Biological Sciences 272, 1153-1161 (2005).
Nielsen, R. Molecular signatures of natural selection. Annual Review of Genetics 39, 197-218 (2005).
Roper, C. et al. Antifolate antimalarial resistance in southeast Africa: A population-based analysis. The Lancet 361, 1174-1181 (2003).
Schneider, K. A. & Kim, Y. An analytical model for genetic hitchhiking in the evolution of antimalarial drug resistance. Theoretical Population Biology 78, 93-108 (2010).
Schneider, K. A. & Kim, Y. Approximations for the Hitchhiking Effect caused by the Evolution of Antimalarial-Drug Resistance. Journal of Mathematical Biology 62, 789-832 (2011).
Vinayak, S. et al. Origin and evolution of sulfadoxine resistant Plasmodium falciparum. PLoS Pathogens 6(3):e1000830 (2010).
Wooton, J. C. et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418, 320-323 (2002).










