


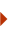

The Conservation track shows a 44-species multiple alignment of vertebrate genomes17 created using the MULTIZ alignment program30 and histograms (wiggle type track) of conservation scores31–33 associated with the alignment (Figure 1, Box I). Conservation tends to peak in coding regions of the gene and fall off in non-coding regions (introns and intergenic regions), so it is a strong signal for a protein-coding gene (Figure 2). Consequently, genomic alignments are used in the prediction of protein-coding genes by programs such as N-SCAN63 and ExoniPhy64 and in the prediction of non-coding RNA genes by programs such as EvoFold.74 These alignments also provide data for tracing genome evolution and for the reconstruction of the ancestral genome sequences for various groups of organisms.
 <i>Genetic Association Database (GAD) View</i>;<sup>38</sup> (B) <i>UCSC</i><sup>15,16,26</sup> <i>Gencode</i>,<sup>54,55</sup> <i>CCDS</i>,<sup>61</sup> <i>RefSeq</i>,<sup>25</sup> <i>MGC</i>,<sup>1–3</sup> and <i>Ensembl</i><sup>18</sup> genes; (C) N-SCAN<sup>63</sup> and ExoniPhy<sup>64</sup> predictions; (D) <i>ACEScan</i>;<sup>75</sup> (E) GenBank <i>Human mRNAs</i> and <i>Spliced ESTs</i>;<sup>27</sup> (F) <i>Poly(A)</i>;<sup>65–67</sup> (G) <i>CpG Islands</i>,<sup>77</sup> <i>GIS ChIP-PET</i>,<sup>71–73,89,90</sup> <i>ORegAnno</i>,<sup>78</sup> and <i>SwitchGear Transcription Start Site (TSS)</i> predictions;<sup>69,70</sup> (H) <i>TargetScan miRNA Regulatory Sites</i> (http://switchgeargenomics.com/);<sup>80,81</sup> (I) <i>Conservation</i>;<sup>17,28–34</sup> and (J) <i>SNPs</i><sup>17,19,35,36</sup> and <i>RepeatMasker</i> repeats (http://www.repeatmasker.org).<sup>37</sup>", "true", "All rights reserved.", '650', '954', 'http://www.nature.com/nature_education');)
Conservation of sequence implies functional significance and can also occur where there are regulatory elements in the genome. Several types of regulatory elements are involved in the control of gene transcription including promoters, enhancers, silencers, insulators, and chromatin structure. Locating these elements is important to further the understanding of the mechanism of gene activation according to environmental conditions, physiology, developmental stages, and tissue type. The human ENCODE project (see the third section of "Gene and Gene Predication Tracks") contributes to the development and use of high throughput experimental techniques to facilitate the discovery of regulatory elements.
The ACEScan75 track predicts conserved alternative exons that are present in some transcripts and skipped by others in both humans and mice (Figure 1, Box D). Enrichment of splicing regulatory motifs occurs in intron regions close to alternative exons, which also show a greater degree of conservation than those close to constitutive exons. ACEScan uses this information to predict the constitutive exon that is skipped in the human mRNA AK129537 and in the UCSC Gene uc010cvx.1.
 5' view; (b) 3' view", "true", "Figure 2", "(A) Conservation is high in the exons in the coding region and falls off at the boundaries of exons. The <i>Conservation</i> track<sup>17</sup> shows two types of conservation scores.<sup>31–33</sup> <i>PhyloP</i> (blue) measures conservation in each alignment column regardless of neighboring columns and it is appropriate for evaluating the selection signatures at particular nucleotides or classes of nucleotides. <i>PhastCons</i> (green) takes into account each column of the alignment and its neighbors to estimate a probability that each nucleotide belongs to a conserved element and thereby results in a smoother plot. (B) In the <i>SNPs (130)</i> track, polymorphisms at the 3' end of <i>PPP1R1B</i> are displayed. In pack or full track display mode, a label to the left of the SNP shows the chimp allele followed by the human alleles.", "true", "All rights reserved.", '650', '867', 'http://www.nature.com/nature_education');)
Transcriptional regulatory elements tend to be enriched near the first exon.76 Evidence of these motifs include the CpG island77 at the 5'end of the PPP1R1B gene locus and the SwitchGear TSS prediction,69,70 which is color-coded according to confidence level (a darker color implies a higher score) (Figure 1, Box G). The ORegAnno78 track displays hand-curated regulatory regions extracted from the literature (Figure 1, Box G). The darker green rectangle represents a regulatory region, located in an intron of the PPP1R1B gene bound by the CCCTC-binding factor (zinc finger protein) (CTCF) transcription factor. The lighter green item below represents the actual location of the CTCF binding site. This binding site was determined by ChIP (chromatin immunoprecipitation) experiments. Details of the method are found by clicking on these track items and following the link to the ORegAnno database record and also the PubMed link for the associated publication. Histone modifications and multiple transcription factor binding sites for a variety of cell types are shown in the GIS ChIP-PET track (the GIS-PET method is described in "GenBank mRNA and EST Sequence Alignments"). Tri-methylation of lysine4 and lysine27 on histone H3 is indicated at the 5' end of the PPP1R1B gene (Figure 1, Box G). Such signals for regulatory elements may be misleading: CpG islands are frequently found in or near promoters of genes but not all genes have them, TSS predictions may contain false positives, and transcription factor binding site measurements can be noisy.
TargetScan79–81 track predicts the presence of a microRNA binding site in a conserved region at the 3' end of the transcript (Figure 1, Box H). The prediction is based partially on conservation, so the TargetScan annotation and conservation are not independent evidence of a regulatory region.
The Conservation track17 shows peaks of conservation that correspond to the coding sequence of the gene (Figure 1, Box I). Conservation falls off at the exon/intron boundaries as illustrated in Figure 2, which show a close-up view of the 5' and 3' ends of the PPP1R1B gene.
At this zoom level, the Conservation track, by default, shows the nucleotide sequence of the aligned genomes and, in coding regions based on the longest UCSC Genes transcript at this locus, the codon translation can be seen for each of the genomes. This enables the user to see not only the conservation at the amino acid level but also where there are differences at the amino acid level between proteins encoded by orthologous protein-coding genes. In Figure 2B, it is possible to see that there is a SNP (rs35797948) in the SNPs (130) track17,36 that is colored red, indicating a non-synonymous mutation in the coding region. Clicking on this SNP displays further information about it and a link to the related entry in dbSNP at NCBI.19,35 This reveals that there are two known alleles (A/G), which code for either an arginine (CGC codon) (as in the reference genome) or a histidine (CAC codon) amino acid in the translation of this last coding exon of PPP1R1B. At neutral pH, both histidine and arginine are positively charged, but histidine is only weakly charged so this is a somewhat conservative substitution. However, histidine's side chain is an aromatic structure, so this type of substitution could potentially disrupt protein structure and/or function. In this case, the substitution occurs close to the C terminus of the protein and, consequently, may have little or no effect on the protein's functionality although this can only be verified with experimental evidence.








