Volume 44 Issue 8, August 2012
Special Issue: NMR of Polymers
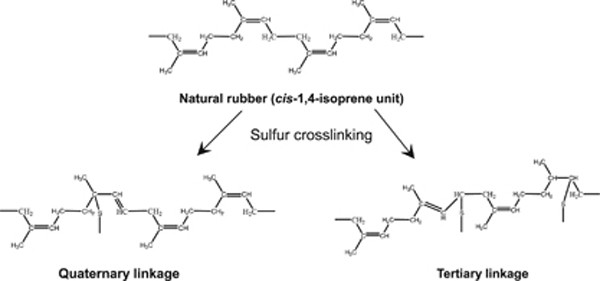
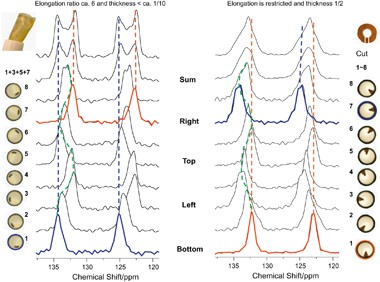
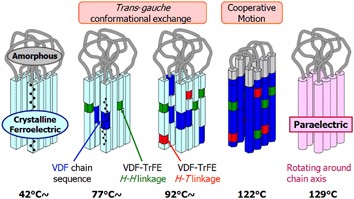
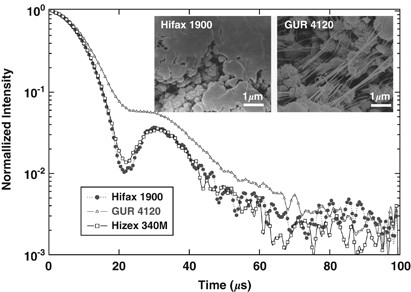
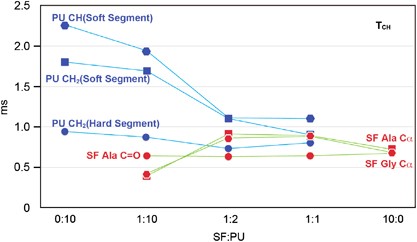
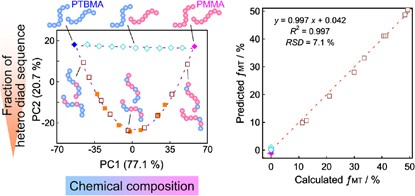
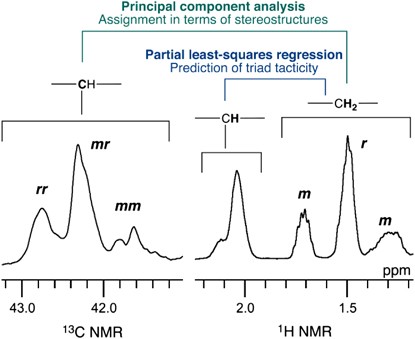
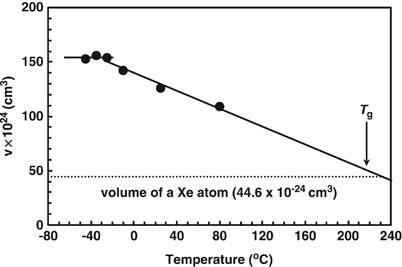
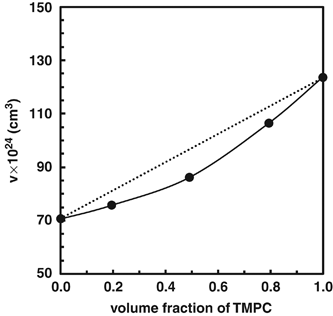
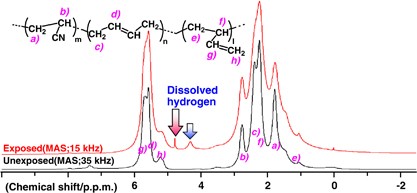
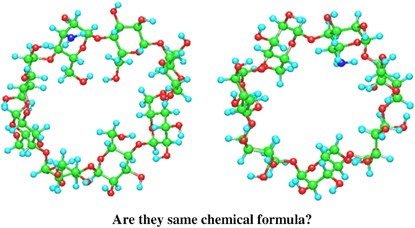
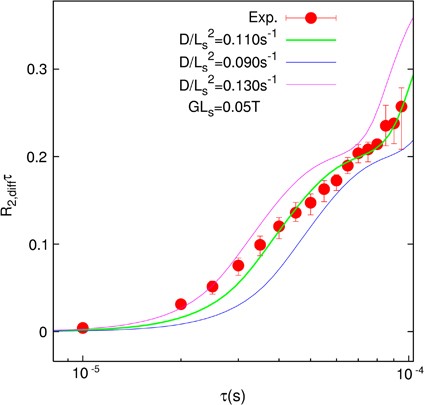
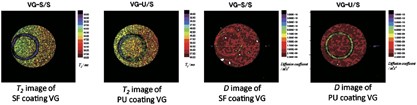
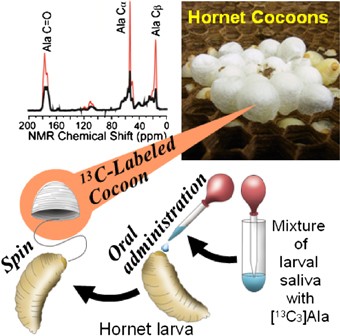
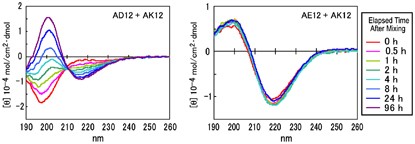
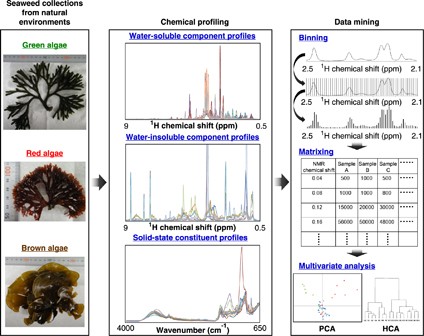
NMR of Polymers: Assorted figures from the papers solicited for the Special Issue and a picture of Superconducting Magnet. Figures (clockwise from top right); Heme coordination structures (Yamamoto), HMBC spectrum of PLA (Suganuma et al.), 129Xe spectra of 129Xe in PPO (Yoshimizu et al.), 13C CPMAS spectra of hornet cocoons (Kameda), 13C spectra and loading histograms of PMMA-co-TBMA (Momose et al.), 1H FID of UHMW-PE reactor powders (Uehara et al.)

Editorial
-
Advertisement