
Abstract
The search for a vaccine against human immunodeficiency virus type 1 (HIV-1) has many hurdles to overcome. Ideally, the stimulation of both broadly neutralizing antibodies and cell-mediated immune responses remains the best option, but no candidate in clinical trials at present has elicited such antibodies, and efficacy trials have not demonstrated any benefit for vaccines designed to stimulate immune responses of CD8+ T cells. Findings obtained with the simian immunodeficiency virus (SIV) monkey model have provided new evidence that stimulating effective CD8+ T cell immunity could provide protection, and in this Perspective we explore the path forward for optimizing such responses in humans.
Similar content being viewed by others

Main
Despite advances in antiretroviral therapy and partial management of the AIDS epidemic by other measures, only a vaccine can end the human immunodeficiency virus type 1 (HIV-1) pandemic. Ideally, such a vaccine would elicit both humoral immune responses and cellular immune responses to prevent infection with HIV-1 and to control such infection, respectively. The vaccine would stimulate broadly neutralizing antibodies, but no candidate immunogen has been able to do that so far. Such antibodies are possible because they are present at low concentrations in ∼20% of patients infected long term with HIV-1, but most are highly somatically mutated and are often derived from germline-encoded antibodies that hardly bind at all to the HIV-1 envelope (Env)1,2. A proportion are also self-reactive, which limits their expansion. Such features make vaccine design very difficult. However, non-neutralizing antibodies may also be effective, and a combination of priming with a vaccine of Env delivered via a canarypox vector followed by boosting with the HIV-1 glycoprotein gp120 provided weak protection (31%) in the RV144 trial3. Non-neutralizing antibodies were stimulated, but although correlates of infection risk were identified4, correlates of protection have not yet been defined. A vaccine against HIV-1 based solely on the elicitation of protective antibodies is still many years away.
Vaccines that stimulate cytotoxic T lymphocytes
A vaccine that stimulates HIV-1-specific CD8+ cytotoxic T lymphocyte (CTL) responses is a possible alternative to the vaccines noted above, at least until a way of stimulating neutralizing antibodies is found. During natural infection with HIV-1, CTLs control but do not eliminate viremia5. In rhesus monkeys, vaccines that stimulate CTLs lead to better than natural control of simian immunodeficiency virus (SIV) or SIV-HIV hybrid viruses after subsequent challenge6 (Fig. 1). However, in the STEP trial, a vaccine based on recombinant adenovirus 5 designed to stimulate CTLs specific for the group-associated antigen (Gag), polymerase (Pol) and negative factor (Nef) proteins of HIV-1 did not improve control of HIV-1 in volunteers who subsequently became infected. Worse, there was increased acquisition of HIV-1 infection in participants in this vaccine trial7. That risk was strongly associated with the presence of preexisting antibodies to the adenovirus vector and lack of circumcision in males; those factors could have over-ridden any weak protection provided by the vaccine. However, when those risk groups were excluded from the HVTN505 trial, which tested a regimen of priming with DNA and boosting with the adenovirus 5–based vaccine, vaccination still provided no protection from infection with HIV-1 (ref. 8). Consequently, these negative vaccine-trial results have raised questions about the whole concept of a CTL-inducing vaccine against HIV-1.

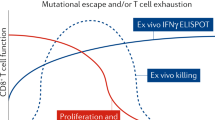
Effect of various T cell–stimulating vaccines (key) on viral load over time (with infection on day 0) during natural infection with HIV or SIV, showing the decrease in viral load achieved without a vaccine (None), by CTL responses (Partial control; as in ref. 6, for example), by the RhCMV vaccine (Slow eradication)9,10 and by a hypothetical vaccine that targets the virus at the site of infection (Rapid eradication).
Now that approach has been reawakened by an exciting new approach9,10. In those studies, a rhesus monkey cytomegalovirus (RhCMV) vector with recombinant SIV genes as the immunogen elicited strong persisting effector memory CTL responses in rhesus monkeys. After being challenged with pathogenic SIVmac239 virus, all vaccinated monkeys were infected, but 50% subsequently cleared virus soon after the peak viremia of acute infection, after systemic spread—an unprecedented event (Fig. 1). In monkeys that cleared the virus, later removal of CD8+ T cells in vivo did not cause a rebound of viremia9,10.
Remarkably, that CMV-based vaccine elicited very atypical T cell responses that were exceptionally broad and even, with a mean of 34 epitopes of Gag alone, compared with 9–10, ranked in immunodominance hierarchies, elicited through the use of conventional vectors11. Such breadth would make escape of the virus very unlikely. The vaccine did not elicit classical immunoprevalent CTL responses, and two-thirds of the CD8+ T cells were restricted by major histocompatibility complex (MHC) class II. Nevertheless, the CTLs recognized SIV-infected cells, which indicates that the same epitopes must be presented naturally. This suggests that the difference between this CMV vector and other vaccines, and SIV itself, is in its priming of such atypical CD8+ T cells. The induction of such very broadly reacting CD8+ T cells requires deletion of three rhCMV genes (equivalent to human CMV genes encoding the entry factors UL128, UL130 and UL131) encoding molecules that determine viral tropism to, for example, dendritic cells. Suppression of the canonical MHC class I–restricted responses of CD8+ T cells was dependent on the presence of the RhCMV equivalent of the human CMV gene encoding US11, which recycles newly synthesized MHC class I molecules for degradation. Thus, abnormal priming of T cells probably occurs via atypical antigen-presenting cells with defective processing of antigens presented by MHC class I.
However, it is not yet clear whether such protection is actually dependent on those atypical CD8+ T cell responses. If they are, it is possible that the vaccine delivered via the RhCMV vector works because SIV has never before encountered MHC class II–restricted CD8+ CTL responses and has not evolved to evade them. However, beyond the breadth of the response, the quantity of the persisting CTLs and their state of activation may be the most critical protective factors. Such issues could be addressed by vaccination with RhCMV vectors that do not have deletion of the genes encoding UL128, UL130 and UL131 and lack the equivalent gene encoding US11. Also, it will be important to determine why only 50% of the monkeys were protected. Given the reproducibility of that finding9,10, there is a clear opportunity here to identify correlates of protection. These critical experiments should determine unequivocally what makes those T cell responses protective and open the way to finding vaccines that elicit similar CD8+ T cell responses in humans.
Viral variability
An issue that is difficult to address adequately with studies of vaccines against SIV is the variability of HIV-1. This impairs vaccine efficacy in two ways. First, there is a high likelihood of a mismatch between the vaccine sequences and the infecting virus. For CTLs, a difference of a single amino acid in an epitope (∼10% variance) has a 30–50% chance of negating recognition by T cells12. Within an HIV-1 subtype, viral sequences vary by 10–20%, with Env, transactivator of transcription (Tat) and Nef being more variable than Gag p24 or Pol. Between clades, the differences are much greater. Second, if an infecting virus is not immediately silenced, there is considerable potential for the virus to escape and for the loss of any vaccine-induced protection13. That probably occurred in the STEP trial, in which there was 'sieving' of epitope sequences by the T cell responses, which could have abrogated weak protective effects14. Given that the HVTN505 trial generated CD8+ T cells that were mostly specific for Env, similar events might have masked weak protection in a subset of subjects.
Two new strategies for a CD8+ T cell–eliciting vaccine against HIV-1 that focus on the immunogen rather than the vector are aimed at addressing those issues (Fig. 2). The first is the 'mosaic' approach15,16,17. In this strategy, the vaccine comprises two or more sequences, administered together. Those are designed from naturally occurring viral sequences but are engineered to maximize perfect coverage of a nine–amino acid segment shared by the most common variants but always include naturally occurring sequence stretches. Thus, a two-mosaic vaccine can cover the two most common alternatives at each variable position. More components would be even better, but the practicalities of vaccine manufacturing limit this to two or three. Such constructs provide much better coverage by testing all possible nine–amino acid sequences for matches compared with those of vaccines containing natural, consensus or ancestral HIV-1 sequences. When administered to monkeys, mosaic vaccines induced stronger, broader (more epitopes) and deeper (greater cross-reactivity, including variations not in the immunogen itself) responses than did single immunogens16,17. However, natural T cell–response immunodominance patterns were seen, with no focus on the more conserved epitopes. Although strong responses to the conserved epitopes were noted, they were often subdominant to variable epitopes18. So, unless nearly complete control of a virus is achieved, the virus could still escape by mutating those variable epitopes, particularly when the responding cells are immunodominant, so that the T cells outcompete those specific for more conserved epitopes13.

Construction of vaccines based on viral sequences in four viral isolates (top; simplified representation): horizontal lines indicate viral sequences; circles indicate sites of greatest variability between isolates (and potential escape mutations from CTL pressure; there may be more than two alternative sequences at each spot); and blue lines indicate regions of relative conservation (although in reality no region of HIV-1 is invariant). The mosaic vaccine (middle) is constructed to include the most common variants from the isolates in as few strands as possible while conserving naturally occurring sequence stretches. In the conserved region–containing vaccine (bottom), the relatively conserved regions (blue) are excised and then are 'stitched' together (which creates an unnatural junctional region). The regions typically vary from 30 to 120 amino acids in length.
The second approach focuses CTLs only on conserved epitopes19,20,21. This could be achieved through the use of vaccines comprising strings of epitopes, but these are poorly immunogenic22, and the diversity of epitopes dictated by genetic polymorphism in HLA molecules and multiple junctions between epitopes that could interfere with T cell responses are confounding factors. Therefore, vaccine candidates comprising only the most conserved regions of the HIV-1 genome have been designed to focus T cell responses on the least variable epitopes, which allows multiple HLA types to select epitopes in the construct19,20. Another group has gone a step further by identifying epitopes associated with good control of the virus in very large patient cohorts21. Many of those epitopes are highly conserved and are in Gag p24 or Pol, so there is overlap between the approaches. Because of residual variability even in 'conserved' regions, the coverage (as defined above) of these immunogens is not quite as good as that of mosaic constructs, but it would be a small step to construct mosaics of the conserved region–containing vaccine(s) and thereby improve the match to any infecting virus and further limit the chances that the virus would escape after infection. A theoretical risk of using such artificial immunogens is that the T cells induced could be specific for epitopes only processed and presented from the construct rather than from natural HIV-1-infected cells. However, human T cells induced by a vaccine containing conserved regions recognize HIV-infected cells and suppress virus replication in vitro23.
Should Env be included in a vaccine that stimulates CTLs?
It is frequently argued that an Env immunogen should be included in any vaccine against HIV to be used in efficacy trials because of the RV144 trial result; the vaccine might generate protective non-neutralizing antibodies. However, Env-specific CTLs have never been shown to be protective, either in the HVTN 505 trial8 or in natural-history studies21,24, probably because of epitope variability. So not only might Env-specific CTLs be ineffective but also, if they were immunodominant, as in the HVTN505 trial8, they might lessen the effectiveness of CTLs specific for conserved epitopes by competing for epitopes on the same infected cells. Although some monkey studies have shown that the inclusion of Env can reduce acquisition of SIV after multiple low-dose challenges, akin to the results obtained in the RV144 trial, the protection only delays infection and has little effect on viral load25. The RhCMV vectors used in the studies noted above9,10 did contain the gene encoding Env together with most other SIV genes, but there was no detectable antibody response, and the CTL responses were so broad and atypical that any positive or negative effects of Env-specific CTLs were probably diluted. Overall, the case for adding Env, in a form that does not induce broadly neutralizing antibodies, to a CTL-inducing vaccine is unconvincing.
Vectors
For the induction of CTL responses, the vaccine vectors used will be as critical as the insert. In humans, adenovirus 5 vectors have induced strong CTL responses7, but uncertainty remains about the increased risk of acquisition of infection. The careful use of vectors that do not cross-react with recurrent human pathogens and that stimulate mainly CD8+ T cells rather than CD4+ T cells is probably the best way forward. Replicating viral vectors with low seroprevalence in the population remain an attractive option because of their potential to mimic the best attributes of successful live attenuated vaccines26. The use of other viral vectors, as well as approaches that incorporate DNA transfected into cells through electroporation, need to be further explored and compared. A human version of the CMV vector that has enabled clearance of SIV in monkeys is an exciting possibility, but ensuring safety (given that CMV is a persistent, replicating, pathogenic human virus) remains paramount and will present regulatory challenges. Numerous strategies to engineer human CMV to address the safety concerns are in development at present. Alternatively, it might be possible to design other vectors, including those that incorporate other herpes viruses, that elicit the same kind of T cell response10,11. Finding correlates of protection in the 50% of RhCMV-vaccinated monkeys that clear SIV could open this route to other vectors.
Selecting vaccines for efficacy trials
If a future vaccine is designed that stimulates the production of broadly neutralizing antibodies in phase I vaccine trials, there would be a strong case to take it to an efficacy trial. It will be more difficult to select candidate vaccines that stimulate protective non-neutralizing antibodies4. Extensions of the RV144 trial are being planned in Thailand, and trials with related products that use more potent adjuvants and additional vectors are planned for initiation in South Africa beginning in 2016, in subjects at high risk for infection with HIV. If efficacy is achieved, these trials might provide greater understanding of the immunological correlates of protection and might provide benchmarks for the future selection of candidates for efficacy trials. However, there is a risk that the initial result may not be repeatable or that protection will be lower in populations at very high risk of infection with HIV-1.
For CTLs, the most likely correlates of protection will be the specificity and breadth of the T cells and their functional activity11,13,21, although these parameters could be refined once more is known about how the vaccine delivered via the CMV vector works. To deal with viral variability, mosaic vaccines or conserved region–containing vaccines delivered by strong vectors or with good adjuvants should generate strong CTL responses to conserved or protective epitopes13,27. The requisite quality of the T cells remains more controversial. The most relevant test is probably suppression of virus-infected cells in vitro28,29, arguably the T cell equivalent of neutralization of the virus by antibodies, although the standardization of panels of viruses to assess breadth of HIV-specific CTL responses in vitro is still in its infancy. Suppression of virus in that assay correlated directly with control of the virus28,30 and correlated inversely with the rate of the decrease in CD4+ T cell numbers in a prospective study of acute infection29. Suppression of virus in vitro and in vivo correlates with antigen sensitivity, the use of T cell antigen receptors and lytic capacity27,31,32. However, the CTLs also must persist, and this may require the use of persisting vectors (exemplified by RhCMV), repeated delivery of immunogens or the use of slowly released immunogens.
Thus, the aim should be to find vaccines that stimulate high CTL responses that are both broad and specific for conserved epitopes, are able to suppress HIV-1 replication in vitro and are sustained for several months. Such vaccines would be identified in phase I trials. Once a suitable candidate is found for efficacy testing, should it be tested alone or in combination with an Env-containing vaccine able to generate broadly neutralizing antibodies or other protective antibodies? If the two approaches are distinct (i.e., the vaccine that elicits CD8+ T cells focuses on conserved or mosaic internal protein epitopes, and the antibody-inducing vaccine elicits Env-specific antibodies with few or no CTLs), the two vaccines should not interfere with each other. One strategy to help accelerate vaccine development would be an adaptive design in which the Env and CTL-eliciting components are tested individually and in combination. However, it is unlikely that both components of the vaccine will be available for testing at the same time. Therefore, if the data warrant advancing a CTL-based vaccine to efficacy trials, the vaccine should not be held back awaiting an appropriate vaccine based on antibodies to HIV Env.
Efficacy trial end points for vaccines that induce CTLs
Until now, CTL-inducing vaccines have been aimed at reducing a set point of the viral load. However, that endpoint will be confounded by the increasing use of early antiretroviral therapy, before the set point is reached, both for treatment and to prevent further transmission33. Also, set point is strongly influenced by HLA type34 and viral virulence35. Therefore, a set point of viral load is an unsatisfactory primary endpoint for vaccine trials.
The results obtained for RhCMV10,11 suggest that acquisition of infection with HIV-1 could be an endpoint, provided enough time is given for the initial viremia to be cleared10,11. Because some blood sampling might hit the viremic peak or a 'blip' of viremia, at least two measurements of viral load would be needed. If incomplete control of the virus were achieved, the viral load in plasma at 6 months or later would be a useful secondary endpoint in those subjects not already on antiretroviral therapy. Similarly, measurement of the 'sieving' effects on viral sequence attributable to vaccine-induced CTL responses might also be useful. More discussion of endpoints is needed, but this should not deter the development of new vaccines that stimulate CTL responses. Such vaccines could yet serve a vital role in the fight to prevent and control infection with HIV-1.
Conclusions
The 'T cell vaccine' approach has always been considered less than ideal and finds few precedents among existing successful vaccines. However, results obtained with SIV models in which infections have been aborted or cleared suggest that such vaccines may have greater value than previously thought. Novel ways of dealing with viral diversity are also encouraging. In summary, vaccines that elicit T cells at a minimum may complement antibody-stimulating vaccines and potentially could offer some protection on their own.
References
Haynes, B.F. & McElrath, M.J. Progress in HIV-1 vaccine development. Curr. Opin. HIV AIDS 8, 326–332 (2013).
Verkoczy, L., Kelsoe, G., Moody, M.A. & Haynes, B.F. Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies. Curr. Opin. Immunol. 23, 383–390 (2011).
Rerks-Ngarm, S. et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361, 2209–2220 (2009).
Haynes, B.F. et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366, 1275–1286 (2012).
McMichael, A.J., Borrow, P., Tomaras, G.D., Goonetilleke, N. & Haynes, B.F. The immune response during acute HIV-1 infection: clues for vaccine development. Nat. Rev. Immunol. 10, 11–23 (2010).
Barouch, D.H. et al. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290, 486–492 (2000).
McElrath, M.J. et al. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet 372, 1894–1905 (2008).
Hammer, S.M. et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N. Engl. J. Med. 369, 2083––2092 (2013).
Hansen, S.G. et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473, 523–527 (2011).
Hansen, S.G. et al. Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013).
Hansen, S.G. et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340, 1237874 (2013).
Lee, J.K. et al. T cell cross-reactivity and conformational changes during TCR engagement. J. Exp. Med. 200, 1455–1466 (2004).
Liu, M.K. et al. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J. Clin. Invest. 123, 380–393 (2013).
Rolland, M. et al. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat. Med. 17, 366–371 (2011).
Korber, B.T., Letvin, N.L. & Haynes, B.F. T-cell vaccine strategies for human immunodeficiency virus, the virus with a thousand faces. J. Virol. 83, 8300–8314 (2009).
Santra, S. et al. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat. Med. 16, 324–328 (2010).
Barouch, D.H. et al. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat. Med. 16, 319–323 (2010).
Stephenson, K.E. et al. Full-length HIV-1 immunogens induce greater magnitude and comparable breadth of T lymphocyte responses to conserved HIV-1 regions compared with conserved-region-only HIV-1 immunogens in rhesus monkeys. J. Virol. 86, 11434–11440 (2012).
Létourneau, S. et al. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS ONE 2, e984 (2007).
Rolland, M., Nickle, D.C. & Mullins, J.I. HIV-1 group M conserved elements vaccine. PLoS Pathog. 3, e157 (2007).
Mothe, B. et al. Definition of the viral targets of protective HIV-1-specific T cell responses. J. Transl. Med. 9, 208 (2011).
Goonetilleke, N. et al. Induction of multifunctional human immunodeficiency virus type 1 (HIV-1)-specific T cells capable of proliferation in healthy subjects by using a prime-boost regimen of DNA- and modified vaccinia virus Ankara-vectored vaccines expressing HIV-1 Gag coupled to CD8+ T-cell epitopes. J. Virol. 80, 4717–4728 (2006).
Borthwick, N. et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol. Ther. 22, 464––475 (2014).
Kiepiela, P. et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13, 46–53 (2007).
Barouch, D.H. et al. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature 482, 89–93 (2012).
Parks, C.L., Picker, L.J. & King, C.R. Development of replication-competent viral vectors for HIV vaccine delivery. Curr. Opin. HIV AIDS 8, 402–411 (2013).
Mothe, B. et al. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS ONE 7, e29717 (2012).
Spentzou, A. et al. Viral inhibition assay: a CD8 T cell neutralization assay for use in clinical trials of HIV-1 vaccine candidates. J. Infect. Dis. 201, 720–729 (2010).
Yang, H. et al. Antiviral inhibitory capacity of CD8+ T cells predicts the rate of CD4+ T-cell decline in HIV-1 infection. J. Infect. Dis. 206, 552–561 (2012).
Freel, S.A. et al. Initial HIV-1 antigen-specific CD8+ T cells in acute HIV-1 infection inhibit transmitted/founder virus replication. J. Virol. 86, 6835–6846 (2012).
Almeida, J.R. et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 204, 2473–2485 (2007).
Berger, C.T. et al. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J. Virol. 85, 9334–9345 (2011).
Shapiro, S.Z. Clinical development of candidate HIV vaccines: different problems for different vaccines. AIDS Res. Hum. Retroviruses 10.1089/aid.2013.0114 (22 November 2013).
Pereyra, F. et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330, 1551–1557 (2010).
Alizon, S. et al. Phylogenetic approach reveals that virus genotype largely determines HIV set-point viral load. PLoS Pathog. 6, e1001123 (2010).
Acknowledgements
Supported by the Medical Research Council UK, the National Institute of Allergy and Infectious Diseases of the US National Institutes of Health, the Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery (UM1 AI 00645) and donors to the International AIDS Vaccine Initiative (including the US Agency for International Development and the Bill & Melinda Gates Foundation). The content is solely the responsibility of the authors and does not necessarily represent the official views of the US National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A.J.M. is a named coinventor of a conserved region–containing vaccine against HIV on a patent owned by Medical Research Council Technology, and the International AIDS Vaccine Initiative (of which W.C.K. is the Chief Scientific Officer) has supported clinical research on such vaccines.
Rights and permissions
About this article
Cite this article
McMichael, A., Koff, W. Vaccines that stimulate T cell immunity to HIV-1: the next step. Nat Immunol 15, 319–322 (2014). https://doi.org/10.1038/ni.2844
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.2844
This article is cited by
-
A longitudinal analysis of immune escapes from HLA-B*13-restricted T-cell responses at early stage of CRF01_AE subtype HIV-1 infection and implications for vaccine design
BMC Immunology (2022)
-
Impact of Endemic Infections on HIV Susceptibility in Sub-Saharan Africa
Tropical Diseases, Travel Medicine and Vaccines (2019)
-
Nonhuman primate models of human viral infections
Nature Reviews Immunology (2018)
-
Computational Design of Epitope-Enriched HIV-1 Gag Antigens with Preserved Structure and Function for Induction of Broad CD8+ T Cell Responses
Scientific Reports (2018)
-
Broad HIV-1 inhibition in vitro by vaccine-elicited CD8+ T cells in African adults
Molecular Therapy - Methods & Clinical Development (2016)
