
Abstract
Lysosomal lipid accumulation, defects in membrane trafficking and altered Ca2+ homoeostasis are common features in many lysosomal storage diseases. Mucolipin transient receptor potential channel 1 (TRPML1) is the principle Ca2+ channel in the lysosome. Here we show that TRPML1-mediated lysosomal Ca2+ release, measured using a genetically encoded Ca2+ indicator (GCaMP3) attached directly to TRPML1 and elicited by a potent membrane-permeable synthetic agonist, is dramatically reduced in Niemann–Pick (NP) disease cells. Sphingomyelins (SMs) are plasma membrane lipids that undergo sphingomyelinase (SMase)-mediated hydrolysis in the lysosomes of normal cells, but accumulate distinctively in lysosomes of NP cells. Patch-clamp analyses revealed that TRPML1 channel activity is inhibited by SMs, but potentiated by SMases. In NP-type C cells, increasing TRPML1's expression or activity was sufficient to correct the trafficking defects and reduce lysosome storage and cholesterol accumulation. We propose that abnormal accumulation of luminal lipids causes secondary lysosome storage by blocking TRPML1- and Ca2+-dependent lysosomal trafficking.
Similar content being viewed by others


Introduction
The lysosome, which serves as the cell's recycling centre, can mediate the hydrolase-based degradation of a variety of biomaterials, including proteins, lipids and membranes1. Most lysosomal storage diseases (LSDs), of which more than 50 are known, are due to the defective activities of specific lysosomal enzymes, which result in the lysosomal accumulation of specific substrates2. For instance, in Niemann–Pick (NP) type A and B diseases (NPA and NPB), sphingomyelins (SMs) accumulate in lysosomes due to insufficient activity of acid SMase (aSMase) (Supplementary Fig. S1)3; similarly, insufficient aSMase activity and SM accumulation occur in NP type C (NPC), where the genetic defect occurs in components of an intracellular cholesterol/lipid trafficking pathway3,4. Interestingly, regardless of the nature of the primary undigested metabolites, common cellular defects are seen in a spectrum of LSDs5. For example, defects in lipid trafficking (the exit of lipids from the late endosome and lysosome) and autophagome–lysosome fusion are commonly observed in almost all sphingolipidoses, including NPA, NPB and NPC2,6. In addition, altered Ca2+ and Fe2+ homoeostasis are also common factors in these LSDs2,5,7. Thus, it is likely that the common trafficking machinery is impaired in LSDs with unrelated origins or primary defects. One intriguing hypothesis is that the primary accumulated materials cause a sort of 'lysosome stress,' which impairs common biochemical and/or signalling pathways in the lysosome, similar to the way unfolded proteins cause 'ER stress' to trigger an 'unfolded protein response'2,5.
Cells that lack mucolipin transient receptor potential (TRP) channel 1 (TRPML1) exhibit lysosomal defects similar to those seen in NP cells: defective autophagosome–lysosome fusion or lysosome reformation, abnormal lipid trafficking and altered Ca2+ and Fe2+ homoeostasis8,9,10,11,12,13,14,15. Furthermore, mutations in the human Trpml1 gene cause mucolipidosis type IV (ML4) LSD, which exhibits membrane trafficking defects and excessive lysosomal storage16,17,18. TRPML1 is a ubiquitously expressed Fe2+ and Ca2+ dually permeable channel predominantly localized in late endosomes and lysosomes (LELs)11,13,19,20,21. TRPML1 is specifically activated by phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2), an LEL-localized low-abundance phosphoinositide14. In addition, both TRPML1-lacking and PI(3,5)P2-deficient cells exhibit defects in LEL-to-Golgi retrograde trafficking and autophagosome–lysosome fusion8,15,22,23, suggesting that the TRPML1-PI(3,5)P2 system represents a common signalling pathway essential for late endocytic trafficking.
Owing to the high degree of similarity in lysosomal defects, we hypothesized that TRPML1–PI(3,5)P2 signalling is compromised in NPs and many other LSDs. In this study, we measured lysosomal Ca2+ release with lysosome-targeted, genetically encoded Ca2+ indicators and found that TRPML1-mediated lysosomal Ca2+ release was compromised in NPC and NPA cells. Patch-clamp analyses demonstrated that TRPML1 channel activity was inhibited by micromolar concentrations of SMs, but potentiated by SM-hydrolysing enzyme SMases. In NPC cells, increasing TRPML1's expression or activity was found to be capable of correcting the trafficking defects and decrease cholesterol accumulation. We conclude that SMs are negative regulators of TRPML1 channels under physiological conditions, and that abnormal accumulation of SMs in the lysosome lumen blocks Ca2+-dependent membrane trafficking by directly inhibiting TRPML1.
Results
Identification of a potent synthetic agonist for TRPML1
A recent high-throughput screen identified 15 small-molecule chemical compounds (SF compounds) that can activate recombinant surface-expressed TRPML321. TRPML1 is primarily localized on the membranes of LELs13. Using whole-endolysosome patch-clamp recordings11,14, we found that several SF compounds also weakly activated whole-endolysosome TRPML1-mediated currents (ITRPML1 or IML1, data not shown). In HEK293T cells transfected with surface-expressed mutant TRPML1 channels (TRPML1-L15L/AA-L577L/AA, abbreviated as ML1-4A9,21), one of the SF compounds, SF-5121, induced small cytosolic Ca2+ ([Ca2+]cyt) increases (measured with Fura-2 ratios; Fig. 1a,b), and activated small but significant inwardly rectifying whole-cell currents (IML1−4A; Fig. 1d).

(a) ML-SA1 induced increases in cytosolic [Ca2+], as measured by the Fura-2 ratio (F340/F380) in HEK293T cells expressing TRPML1-L15L/AA-L577L/AA (ML1-4A) at low pH (pH 4.6) external solution. SF-51 (10 μM) induced much smaller responses in the same cells. (b) SF-51 (30 μM) induced increases in cytosolic [Ca2+] in ML1-4A-expressing HEK293T cells at low pH external solution. (c) Chemical structures of SF-51 and ML-SA1. The difference between SF-51 and ML-SA1 is highlighted in red. (d) Activation of whole-cell currents by SF-51 and ML-SA1 in ML1-4A-expressing HEK293T cells. ML1-4A-mediated current (IML1−4A) was elicited by repeated voltage ramps (−140 to +140 mV; 400 ms) with a 4-s interval between ramps. Only a portion of the voltage protocol is shown; holding potential=0 mV. (e) Activation of whole-endolysosome IML1 by ML-SA1 and PI(3,5)P2. Enlarged vacuoles/endolysosomes were isolated from vacuolin-treated HEK293 cells stably expressing ML1-4A. ML-SA1 and PI(3,5)P2 were applied to the cytosolic sides of the isolated vacuoles. The pipette (luminal) solution was a standard external (Tyrode's) solution adjusted to pH 4.6; the bath (internal/cytoplasmic) solution was a K+-based solution (140 mM K+-gluconate). Note that the inward current indicates cations flowing out of the endolysosome. (f) Synergistic activation of whole-endolysosome IML1 by ML-SA1 and PI(3,5)P2 in human fibroblasts. IML1 was activated by PI(3,5)P2 (1 μM) and ML-SA1 (10 μM); coapplication of PI(3,5)P2 and ML-SA1 further increased IML1. (g) ML-SA1 activated endogenous whole-endolysosome ML-like currents (IML−L) in CHO cells. (h) ML-SA1 activated whole-endolysosome IML−L in mouse primary macrophages. (i) ML-SA1 activated whole-endolysosome IML1 in WT (ML1+/+), but not ML4 (ML1−/−), human fibroblasts. For panels a and b, the responses were averaged for 20–40 cells from one representative experiment; dara are presented as the mean±s.e.m.
By conducting a Fura-2-based low-throughput screen for more potent TRPML1 agonists, we identified a SF-51-related compound (Fig. 1c for the chemical structures; Mucolipin Synthetic Agonist 1 or ML-SA1) that could induce significant [Ca2+]cyt increases in HEK293 cells stably or transiently expressing ML1-4A (Fig. 1a). In electrophysiological assays, ML-SA1 robustly activated whole-cell IML1−4A (Fig. 1d) and whole-endolysosome IML1 (Fig. 1e). ML-SA1 also activated whole-cell ITRPML2 and ITRPML3, but not six other related channels (Supplementary Fig. S2). ML-SA1 (10 μM) activation of whole-endolysosome IML1 was comparable to the effect of the endogenous TRPML agonist PI(3,5)P2 (1 μM; Fig. 1e), and these agonists were synergistic with each other (Fig. 1f). ML-SA1 activated an endogenous whole-endolysosome TRPML-like current (IML−L) in all mammalian cell types that we investigated, including Chinese Hamster Ovary (CHO) (Fig. 1g), Cos-1, HEK293, skeletal muscle, pancreatic β and macrophage cells (Fig. 1h). ML-SA1 activated whole-endolysosome IML−L in wild-type (WT; ML1+/+), but not ML4 (ML1−/−) human fibroblasts (Fig. 1i), suggesting that although ML-SA1 targets all three TRPMLs, the expression levels of TRPML2 and TRPML3 are very low, and TRPML1 is the predominant lysosomal TRPML channel in this cell type. Thus, we have identified a reasonably specific and potent agonist that can be a useful chemical tool for studying the functions of TRPMLs, especially TRPML1.
A lysosome-targeted genetically encoded Ca2+ indicator
To measure lysosomal Ca2+ release in intact cells, we engineered GCaMP3, a single-wavelength genetically encoded Ca2+ indicator24, to the cytoplasmic amino terminus of TRPML1 (Fig. 2a). When transfected into HEK293T, CHO or Cos-1 cell lines, GCaMP3-TRPML1 (GCaMP3-ML1) was mainly localized in the Lamp-1 and LysoTracker-positive compartments, that is, LEL (Fig. 2b,c). To determine the specificity of the GCaMP3-ML1 fluorescence signal, we tested a variety of Ca2+ release reagents and found that GCaMP3 fluorescence (F470) responded preferentially and reliably to lysosome-acting Ca2+-mobilization reagents (ΔF/F0=0.2–1), including glycyl-L-phenylalanine 2-naphthylamide (GPN, a Cathepsin C substrate that induces osmotic lysis of lysosomes; Fig. 2d) and bafilomycin A1 (Baf-A1, a V-ATPase inhibitor)25. In a subset of cells, GCaMP3 also responded to large (ΔF/F0>2) Ca2+ increases triggered by thapsigargin (targeting the endoplasmic recticulum (ER) Ca2+ store; Fig. 2d), which may presumably spread to close proximity of lysosomes. In the majority of the cells, however, TG failed to significantly increase GCaMP3 fluorescence (Fig. 2d), suggesting that small ER Ca2+ release was not detected by GCaMP3-ML1. Taken together, GCaMP3-ML1 readily and preferentially detects juxta-lysosomal Ca2+ increases.

(a) GCaMP3–TRPML1 (GCaMP3–ML1) fusion strategy. GCaMP3 is fused to the N-terminus of TRPML1. (b) Colocalization of GCaMP3-ML1 fluorescence with Lamp-1 in HEK293T cells expressing both GCaMP3-ML1 and Lamp-1-mCherry. Scale bar=5 μm. (c) Colocalization of GCaMP3-ML1 fluorescence with LysoTracker in GCaMP3-ML1-expressing HEK293T cells. Scale bar=2 μm. (d) Preferential detection of Ca2+ release from lysosome stores by GCaMP3-ML1 in GCaMP3-ML1-transfected Cos-1 cells. ER and lysosome Ca2+ releases were triggered by thapsigargin (TG, 2 μM) and GPN (200 μM), respectively, in low (nominally free Ca2+ + 1 mM EGTA; free [Ca2+] estimated to be <10 nM) external [Ca2+]. All cells responded to GPN; only one out of four cells responded to TG.
In GCaMP3-ML1-transfected CHO (Fig. 3a,b), HEK293T and Cos-1 cell lines, application of ML-SA1 (20 μM) induced rapid increases in GCaMP3 fluorescence (ΔF/F0=0.45±0.05, n=77 cells; Fig. 3a,k) in the presence of low external Ca2+ (nominally free Ca2+ + 1 mM EGTA; free Ca2+ estimated to be <10 nM). By contrast, no significant increase in GCaMP3 fluorescence (ΔF/F0=0.03±0.01, n=32 cells; Fig. 3c,d and k) was observed in cells transfected with a plasmid carrying GCaMP3 fused to a non-conducting pore mutant of TRPML18,14 (GCaMP3-ML1-KK). Both GPN and Baf-A1 largely abolished ML-SA1-induced Ca2+ responses (Fig. 3e–k). By contrast, ML-SA1-induced Ca2+ response was mostly intact in TG-treated cells (Fig. 3k). Collectively, these results suggest that ML-SA1 activated endolysosomal TRPML1 to induce Ca2+ release from lysosomes. Thus, we have established a robust Ca2+ imaging assay to measure TRPML1-mediated lysosomal Ca2+ release.

(a) ML-SA1 (20 μM) induced rapid increases in GCaMP3 fluorescence (measured as change of GCaMP3 fluorescence ΔF over basal fluorescence F0; ΔF/F0) under low (< 10 nM) external Ca2+ in CHO cells transfected with GCaMP3-ML1. Subsequent application of GPN (200 μM) induced smaller responses. Maximal responses were induced by ionomycin (1 μM) application. (b) Representative micrographs from panel (a) to show the changes of GCaMP3 fluorescence upon bath application of ML-SA1 and ionomycin to GCaMP3-ML1-transfected CHO cells. Scale bar=5 μm. (c) ML-SA1 failed to induce significant Ca2+ increases in CHO cells transfected with GCaMP3-ML1-KK (non-conducting pore mutation). (d) Basal GCaMP3 fluorescence (normalized to the maximal ionomycin-induced fluorescence) of GCaMP3-ML1-KK and GCaMP3-ML1. (e) GPN (200 μM) pretreatment abolished ML-SA1-induced responses in GCaMP3-ML1-expressing CHO cells. (f) Repetitive applications of ML-SA1 (20 μM) induced little or no responses in GCaMP3-ML1-expressing CHO cells. (g) Repetitive applications of GPN (200 μM) induced smaller GCaMP3 responses in GCaMP3-ML1-expressing CHO cells. (h) ML-SA1 failed to further increase GCaMP3 fluorescence in GCaMP3-ML1-expressing CHO cells in the presence of GPN (200 μM). (i) ML-SA1 induced small responses in GCaMP3-ML1-expressing CHO cells that had received an application of GPN. (j) Baf-A1 (500 nM) pretreatment abolished ML-SA1-induced Ca2+ response in GCaMP3-ML1-expressing CHO cells. (k) ML-SA1-induced responses in CHO cells transfected with GCaMP3-ML1-KK (non-conducting pore mutation), and GCaMP3-ML1-transfected cells pretreated with Baf-A1, GPN or TG. For panels d and k, the responses were averaged for 40–100 transfected cells from at least three independent experiments; data are presented as the mean±s.e.m. Statistical comparisons were made using analysis of variance: *P<0.05.
Reduced TRPML1-mediated lysosomal Ca2+ release in NPC cells
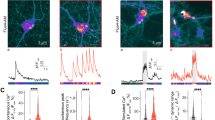
Next, we investigated whether TRPML1-mediated lysosomal Ca2+ release is altered in NP cells. We chose an easy-to-transfect cellular model of NPC, NPC (NPC1−/−) CHO cells, which exhibit all the hallmark features of NPC disease26,27. WT CHO cells transfected with GCaMP3-ML1 exhibited large TRPML1-mediated lysosomal Ca2+ release (Fig. 4a,l). In contrast, NPC CHO cells exhibited much smaller TRPML1-mediated Ca2+ release with a ~75% reduction compared with WT controls (Fig. 4b,l). GCaMP3-ML1 was still mainly colocalized with Lamp-1 in NPC cells (Fig. 4c). In addition, the expression levels of GCaMP3-ML1, as estimated from the maximal fluorescence intensity induced by ionomycin, were comparable for WT and NPC cells (Fig. 4d). A previous study reported that lysosomal Ca2+ stores are compromised in NPC cells25. However, we found that GPN-induced GCaMP3-ML1 increases were comparable for WT and NPC cells (Fig. 4e). Likewise, GPN-induced and NH4Cl-induced Fura-2 responses were also comparable in non-transfected WT and NPC cells (Fig. 4f,g). Furthermore, TRPML1-mediated, but not GPN-induced lysosomal Ca2+ release, was also reduced in GCaMP3-transfected NPC human fibroblasts compared with WT cells (Fig. 4h). Collectively, these results suggest that TRPML1-mediated lysosomal Ca2+ release is selectively reduced in NPC cells.

(a,b) ML-SA1-induced lysosomal Ca2+ release in GCaMP3-ML1-transfected WT (a) and NPC (b) CHO cells. (c) Colocalization of GCaMP3-ML1 fluorescence with Lamp-1 in NPC CHO cells doubly transfected with GCaMP3-ML1 and Lamp-1-mCherry. Scale bar=5 μm. (d) Comparable expression levels of GCaMP3-ML1 in WT and NPC CHO cells. Expression level of GCaMP3-ML1 was estimated by the maximal GCaMP3 fluorescence signal induced by ionomycin (1 μM). (e) Comparable GPN-induced GCaMP3 responses in WT, NPC or U18666A-treated GCaMP3-ML1-transfected CHO cells. (f) NH4Cl (10 mM) and GPN (200 μM) induced comparable levels of Ca2+ increases (measured with Fura-2 ratios) in WT, NPC and U18666A-treated CHO cells. (g) NH4Cl (10 mM) and GPN (200 μM) induced comparable levels of Fura-2 Ca2+ responses in WT and NPC human fibroblasts. (h) TRPML1-mediated, but not GPN-induced, lysosomal Ca2+ release was reduced in GCaMP3-ML1-transfected NPC human fibroblasts compared with WT cells. (i) WT CHO cells were treated with U18666A (2 μg ml−1) for 16 h, and then subjected to filipin staining. Scale bar=10 μm. (j) SMs were stained with Lysenin (0.1 μg ml−1) in WT CHO cells, WT CHO cells treated with U18666A (2 μg ml−1) for 16 h and NPC CHO cells. Scale bar=10 μm. (k) Ca2+ responses in GCaMP3-ML1-transfected WT CHO cells treated with U18666A (2 μg ml−1) for 16–20 h. (l) ML-SA1-induced peak GCaMP3 responses were reduced in GCaMP3-ML1-transfected NPC or U18666A-treated WT CHO cells. (m,n) Endogenous TRPML1-mediated lysosomal Ca2+ release (measured with Fura-2 ratios) in WT (m) and NPC (n) CHO cells. (o) Average endogenous ML-SA1-induced lysosomal Ca2+ responses in WT and NPC CHO cells, and WT, NPC1−/− and ML1−/− mouse macrophages. For panels d, e, f, g, h, k, l and o, the results were averaged for 40–100 cells from at least three independent experiments; data are presented as the mean±s.e.m. Statistical comparisons were made using analysis of variance: *P<0.05.
NPC-like cellular phenotypes can be pharmacologically induced with a small-molecule drug U18666A28. After incubating GCaMP3-ML1-transfected WT CHO cells with U18666A (2 μg ml−1) for 16–20 h, cells exhibited significant increases in cholesterol level as detected by filipin staining4 (Fig. 4i), and sphingomyelin (SM) level as detected by lysenin staining (Fig. 4j). U18666A-treated cells exhibited a ~70% reduction in TRPML1-mediated lysosomal Ca2+ release (Fig. 4k,l). Conversely, drug treatment failed to result in noticeable changes in GPN-induced GCaMP3 responses (Fig. 4e) or Fura-2 Ca2+ responses (Fig. 4f). Taken together, these results suggest a specific reduction of TRPML1-mediated lysosomal Ca2+ release in pharmacologically induced NPC cells.
We next investigated the effect of NPC on endogenous Ca2+ release using Fura-2 Ca2+ imaging. In WT CHO cells, small but significant Ca2+ increases were seen upon application of 100 μM ML-SA1 in low external Ca2+ (ΔFura-2=0.08±0.01, n=89; Fig. 4m,o). In contrast, in NPC CHO cells, ML-SA1 failed to induce significant Ca2+ responses (ΔFura-2=0.01±0.002, n=61; Fig. 4n,o). Cyclodextrin, a polysaccharide that reportedly alleviates NPC cellular phenotypes4, was found to partially restore ML-SA1-induced lysosomal Ca2+ release (Supplementary Fig. S3). Finally, NPC1−/− mouse macrophages, one of the major cell types affected in NPC3, also exhibited a significant reduction in ML-SA1-induced lysosomal Ca2+ release compared with WT cells (Fig. 4o and Supplementary Fig. S4). The ML-SA1-induced lysosomal Ca2+ responses are most likely mediated by TRPML1, as demonstrated by the lack of response in ML1−/− macrophages (Fig. 4o and Supplementary Fig. S4). Collectively, the reduction in TRPML1-mediated lysosomal Ca2+ release may be a general feature of most NPC cell types.
Reduced TRPML1-mediated currents in NPC cell lysosomes
We next investigated the effect of NPC on whole-endolysosome IML1 in human fibroblasts. Compared with WT fibroblasts (Fig. 5a,c), much smaller ML-SA1-activated IML1 was seen in NPC fibroblasts at three different concentrations of ML-SA1 (Fig, 5b,c). On average, whole-endolysosome IML1 activated by 25 μM ML-SA1 was 75±25 pA/pF (n=8) for NPC cells, a ~70% reduction from the values observed for WT cells (442±150 pA/pF; n=6). Notably, the messenger RNA expression levels of TRPML1-3 were not decreased in NPC cells (Fig, 5d). These results suggest that a reduction of whole-endolysosome IML1 accounts for the reduced TRPML1-mediated Ca2+ release observed in NPC cells.

(a,b) Concentration-dependent activation of whole-endolysosome IML−L in WT (a) and NPC (b) human fibroblasts. ML-SA1 (3, 10 and 25 μM) was applied to the cytosolic sides of the vacuoles isolated from human fibroblasts. (c) Average current densities (pA/pF) of IML−L were reduced in NPC fibroblasts compared with WT cells. The current density was calculated from the current size (pA) normalized with the capacitance of the vacuole (pF). (d) Reverse transcriptase PCR analysis of TRPML1-3 messenger RNA expression in WT and NPC human fibroblasts, and NPC1−/− mouse macrophages. Expression levels were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (human fibroblast) and L32 (mouse macrophage). For panels c and d, data are presented as the mean±s.e.m. (WT (blue), n=6; NPC (red), n=8). Statistical comparisons were made using analysis of variance: *P<0.05.
Inhibition of TRPML1 by SMs
The reduced TRPML1-mediated lysosomal Ca2+ release and IML1 in the presence of normal TRPML1 expression in NPC cells suggest tonic inhibition of TRPML1 by cytoplasmic and/or luminal factors. One possibility is that lipids accumulated in NPC lysosomes, such as cholesterol, SM, sphingosine and glycosphingolipids4,29, might directly inhibit TRPML1. To test this possibility, we performed whole-cell recordings in ML1-4A-expressing HEK293T cells. Under this configuration, the extracellular side is analogous to the luminal side (Supplementary Fig. S1), making it possible to study the effects of luminal lipids on IML1−4A. SMs, but not cholesterol or glycosphingolipids (data not shown), acutely inhibited SF-51- (Fig, 6a,d) or ML-SA1-activated IML1−4A. In contrast, SMs failed to inhibit IML1−Va, a gain-of-function mutation that locks TRPML1 in the open state13 (Fig. 6b,d). SMs can be hydrolysed into ceramide and phosphocholine by SMase (Supplementary Fig. S1). No significant inhibition was seen with ceramide (Fig. 6c,d) or phosphocholine (Fig. 6d). Sphingosine, a hydrolysed product of ceramide, potentiated IML1−4A (Fig. 6e).

(a) SF-51-activated whole-cell IML1−4A was inhibited by SMs (20 μM) at low extracellular pH in ML1-4A-expressing HEK293T cells. (b) Insensitivity of whole-cell IML1−Va to SMs (60 μM). (c) Lack of effect of ceramide (Cer, 20 μM) on whole-cell IML1−4A. (d) Summary of the effects of various sphingolipids on whole-cell IML1−4A in ML1-4A-expressing HEK293T cells. PC: phosphocholine. (e) SF-51-activated whole-cell IML1−4A was potentiated by sphingosine (Sph; 5 μM) at low external pH in ML1-4A-expressing HEK293T cells. (f) The effects of SMase (50 ng ml−1) treatment on IML1−4A at low external pH. IML1−4A was elicited by repeated voltage ramps (−140 to + 140 mV; 400 ms) with a 4-s interval between ramps. Current amplitudes at −140 mV were used to plot the time dependence. (g) Representative traces of IML1−4A before (black) and after (dark yellow) SMase treatment at two different time points, as shown in f. (h,i) SMase (50 ng ml−1) treatment alone had no (six out of seven cells; see h for an example) or small (one out of seven cells; i) activation effect on basal whole-cell IML1−4A in ML1-4A-expressing HEK293T cells. (j) The effects of SMase (50 ng ml−1) treatment on SF-51-activated IML1−4A at neutral external pH (pH 7.4). (k) The potentiation effect of SMase was more dramatic at pH 4.6, but abolished by heat inactivation of the enzyme activity. HI-SMase: heat-inactivated SMase. For panels d and k, data are presented as the mean±s.e.m.; the n numbers are in parentheses. Statistical comparisons were made using analysis of variance: *P<0.05.
The robust inhibition of IML1−4A by SMs, but not ceramide or phosphocholine, suggests that the activity of SMase may regulate IML1−4A. Indeed, bath application of SMases, but not heat-inactivated enzymes, significantly potentiated SF-51-activated IML1−4A (Fig. 6f–k), with the potentiation being more dramatic at acidic extracellular (luminal) pH than at neutral pH (Fig. 6k).
SM accumulation reduces TRPML1-mediated Ca2+ release
The results presented above suggest that SM-mediated inhibition underlies NPC-mediated reduction in TRPML1-mediated lysosomal Ca2+ release. Consistent with this, in fibroblasts derived from NPA patients, which completely lack the activity of aSMase3, TRPML1-mediated, but not GPN-induced, lysosomal Ca2+ release (Fig. 7a–c) was significantly reduced. To further test this hypothesis, we induced lysosomal SM accumulation by incubating cells with desipramine and fluoxetine for 2–4 h, which cause proteolytic degradation of aSMase30,31. Compared with non-treated cells, TRPML1-mediated lysosomal Ca2+ release was dramatically reduced in desipramine/fluoxetine-treated CHO cells, and the reduction was abolished in the presence of a protease inhibitor leupeptin30,31 (Fig. 7d–g). Taken together, these results suggest that SM inhibition of TRPML1 underlies NPA- or NPC-mediated attenuation of TRPML1-mediated lysosomal Ca2+ release.

(a,b) ML-SA1-induced lysosomal Ca2+ release was reduced in GCaMP3-ML1-transfected NPA fibroblasts (b) compared with WT fibroblasts (a). (c) Average ML-SA1- (20 μM) and GPN-induced (200 μM) responses in GCaMP3-ML1-transfected WT and NPA fibroblasts. (d) Desipramine (25 μM) treatment for 2–4 h reduced TRPML1-mediated lysosomal Ca2+ release responses (20 μM ML-SA1) in GCaMP3-ML1-transfected CHO cells. (e) Pretreatment of a protease inhibitor leupeptin (25 μM) abolished the effects of desipramine in ML-SA1-induced GCaMP3 responses. (f) Summary of the effects of desipramine (25 μM) and fluoxetine (25 μM) treatment for 2–4 h on TRPML1-mediated lysosomal Ca2+ release responses (20 μM ML-SA1) in GCaMP3-ML1-transfected CHO cells. (g) Desipramine and fluoxetine treatment for 2–4 h reduced endogenous lysosomal Ca2+ release responses (measured with Fura-2 ratios; 100 μM ML-SA1) in CHO cells. For panels c, f and g, the results were averaged for 40–100 cells from at least three independent experiments; data are presented as the mean±s.e.m. Statistical comparisons were made using analysis of variance: *P<0.05.
TRPML1 regulates lysosomal lipid and cholesterol trafficking
Human NPC, NPA and NPB fibroblasts exhibit defects in LEL-to-Golgi trafficking of lipids and proteins6,27. Consistent with this, BODIPY-LacCer, a fluorescent analogue of lactosylceramide that is localized in the Golgi apparatus of WT cells after a pulse-chase period32, was predominantly localized in the LEL-like compartments of NPC1−/− mouse macrophages (Fig. 8a). Owing to the established roles of PI(3,5)P2/TRPML1/Ca2+ in LEL-to-Golgi lipid trafficking15, it is possible that the trafficking defects seen in NP cells are due to the aforementioned attenuated TRPML1-mediated lysosomal Ca2+ release. If so, one would expect that boosting TRPML1 activity may correct the trafficking defects. Indeed, more than 60% of ML-SA1-treated NPC macrophages exhibited a Golgi-like pattern of LacCer staining, suggestive of a significant restoration in LEL-to-Golgi transport (Fig. 8a). In NPC1−/− mouse macrophages and TRPML1-transfected NPC CHO cells, the puncta number of LacCer staining, that reflects the function of LEL-to-Golgi transport, was significantly reduced in the presence of ML-SA1 (Fig. 8b and Supplementary Fig. S5). Collectively, these results suggest that increasing TRPML1's channel activity may restore the trafficking defects in NPC cells, and that TRPML1 deregulation is a pathogenic mechanism underlying lipid trafficking defects in NP cells.

(a) A fluorescent analogue of lactosylceramide, BODIPY-LacCer, was mainly localized in the Golgi-like structures of WT macrophages after a pulse-chase for 1 h, but accumulated in the LEL-like vesicles of NPC macrophages. ML-SA1 (10 μM) treatment of NPC1−/− macrophages for 12 h resulted in a primary Golgi-like localization for LacCer. Scale bar=10 μm. (b) ML-SA1 reduced the number of LacCer puncta in NPC1−/− macrophages. (c,d) ML-SA1 reduced unesterified cholesterol (filipin) staining in ML1, but not ML1-KK-expressing U18666A-treated WT CHO cells. Differential interference contrast (DIC) images are shown for comparison. Scale bar=10 μm. (e) Unesterified cholesterol (filipin) staining in NPC CHO cells was partially reduced in cells expressing DsRed-aSMase. (f) Average filipin staining (intensity normalized to non-transfected control cells) in DsRed-aSMase-transfected cells. (g) Unesterified cholesterol (filipin) staining in NPC CHO cells was partially reduced in cells expressing ML1-Va-EGFP (ML1Va). (h) Average filipin staining (intensity normalized to non-transfected control cells) in ML1Va-transfected cells. Scale bar=10 μm for panels a, c, e and g. For panels b, d, f and h, the results were averaged for multiple randomly taken micrographs from at least three independent experiments. Data are presented as the mean±s.e.m. Statistical comparisons were made using analysis of variance: *P<0.05.
NPC cells accumulate cholesterol, as demonstrated by elevated filipin staining25. However, in U18666A-treated CHO cells, ML-SA1 induced a significant reduction in filipin staining in TRPML1, but not TRPML1-KK-transfected cells (Fig. 8c,d). Likewise, in TRPML1-transfected NPC CHO cells, a significant reduction of filipin staining was also seen in the presence of ML-SA1 (Supplementary Fig. S5). Reduction of filipin staining was also seen in aSMase- (Fig. 8e,f) and TRPML1Va-transfected NPC CHO cells (Fig. 8g,h). These results suggest that increased TRPML1 activity may reduce cholesterol accumulation in NPC cells.
Discussion
We used a lysosome-targeted GCaMP3-based Ca2+ imaging method to demonstrate that TRPML1-mediated lysosomal Ca2+ release, but not the lysosomal Ca2+ store as has been reported previously25, is selectively compromised in NPC and NPA cells. SM, a sphingolipid that is known to accumulate in all three types of NP cells4, inhibits TRPML1. Pharmacologically induced SM accumulation significantly reduces TRPML1-mediated lysosomal Ca2+ release. Conversely, increasing TRPML1's expression or activity is sufficient to correct the trafficking defects and reduce cholesterol accumulation in NPC cells. Collectively, our work provides a feedback mechanism for luminal lipids to control lipid efflux from the lysosome, as well as a pathogenic cause for the secondary lysosome storage present in many LSDs.
SMs are present on the outer leaflet of the plasma membrane33, suggesting that SM inhibition of TRPML1 might serve as a physiological mechanism to prevent this lysosome channel from being active at the wrong subcellular location, that is, the plasma membrane. For normal cells, SMs in the lysosome would be rapidly degraded by aSMase activity33. In NP cells, however, SMs accumulate in the lumen and limited lysosomal membranes (Supplementary Fig. S1). Although the primary storage is likely to be cholesterol, NPC cells are found to have a much lower level of aSMase activity, and a restoration of aSMase activity could revert cholesterol accumulation34. It is not known whether the level of SM is perturbed in other sphingolipidoses. However, rather than absolute SM levels, the relative localization (plasma membrane versus lysosome) is the factor critical for lysosomal Ca2+ release. In addition, other offending metabolites might also inhibit TRPML1, and/or PI(3,5)P2-metabolizing enzymes. Thus, impaired Ca2+ release might be a common pathogenic mechanism for many LSDs. Although TRPML1 is the dominant lysosomal TRPML channel in most cell types, in other cell types where TRPML2 and TRPML3 are more abundantly expressed13, our proposed SM-mediated inhibitory mechanism may also be applied to TRPML2/3, causing lysosomal trafficking defects. Finally, SM and cholesterol accumulation might also contribute to NP phenotypes via mechanisms unrelated to lysosomal trafficking and TRPMLs3.
LEL-to-Golgi trafficking defects are seen in PI(3,5)P2-deficient cells22, ML4 fibroblasts15,35 and NP fibroblasts, as well as fibroblasts from several other sphingolipid LSDs6,36. Our results suggest that reduced TRPML1-mediated lysosomal Ca2+ release directly causes trafficking defects. We previously reported that impaired Fe2+ homoeostasis facilitates the formation of autofluorescent lipofuscin in ML4 cells11. Interestingly, lipofuscin formation is also increased in NPC cells (Supplementary Fig. S6). For most LSDs, impaired lysosomal degradation due to a lack of hydrolytic enzymes leads to trafficking defects and lysosome storage. However, lysosome storage can in turn affect lysosomal degradation and membrane trafficking, which results in a positive feedback loop and a vicious cycle2. Our work suggests a novel concept that lysosome enhancement by manipulating TRPML1 to speed membrane trafficking may break the vicious cycle, providing a novel therapeutic approach for NPs and many other LSDs.
Methods
Molecular biology and biochemistry
The GCaMP3-ML1 construct was made by inserting the full-length GCaMP3 sequence24 between the HindIII and BamHI sites of a pcDNA6 plasmid that contains the mouse TRPML1 complementary DNA at the XhoI site. TRPML1-L15L/AA-L577L/AA (abbreviated as ML1-4A) and the TRPML1 non-conducting pore mutant (D471K/D472K; abbreviated as ML1-KK) were constructed using a site-directed mutagenesis kit (Qiagen). All constructs were confirmed by sequencing, and protein expression was verified by western blot. CHO, HEK293T, Cos-1 and human fibroblast cells were transiently transfected with GCaMP3-ML1 for electrophysiology, Ca2+ imaging and confocal imaging.
PCR primers used for real-time quantitative PCR analysis of TRPML expression in human fibroblasts and mouse macrophages are as follows:
Human TRPML1: F, 5′-CGGATGACACCTTCGCAGCCTAC-3′; R, 5′-ACGCATACCGGCCCAGTGACAC-3′
Human TRPML2: F, 5′-GGTCACCACACAGCTTGTTCGT-3′; R, 5′-TGATGATACTGATTAATAGCA-3′
Human TRPML3: F, 5′-CCAGAAATTGAAACTGAGTGTT-3′; R, 5′-ATGTTATAGTCAGAGTAAAGTC-3′
Mouse TRPML1: F, 5′-AAACACCCCAGTGTCTCCAG-3′; R, 5′-GAATGACACCGACCCAGACT-3′
ML4 and NPC cell lines
NPC (CT43; NPC1−/−) and WT control (RA25) CHO cells were a gift from Dr Chang TY (Dartmouth Medical School). Human skin fibroblast cells from an ML4 patient (ML1−/−, clone GM02048), an NPC patient (NPC1−/−, clone GM03123) and an NPA patient (clone GM00112), and non-disease control cells (ML1+/+/ NPC1−/−, clone GM00969) were obtained from the Coriell Institute for Medical Research (NJ, USA). Human fibroblasts were transiently transfected by electroporation (260 V, 950 μF) with 100 μg of GCaMP3-ML1 plasmid, and then used for Ca2+ imaging experiments 24 h after electroporation.
Mouse lines
NPC1 KO mice (BALB/cNctr-Npc1m1N/J) and WT littermates were ordered from the Jackson Laboratories. TRPML1 knockout mice were kindly provided by Dr. Susan Slaugenhaupt (Harvard Medical School) and Dr. Jim Pickel (NIH)37. Animals were used under approved animal protocols and University of Michigan Institutional Animal Care Guidelines.
Preparation and culture of mouse macrophages
Bone marrow cells from femurs and tibias were harvested and cultured in macrophage differentiation medium (RPMI-1640 medium with 10% fetal bovine serum (FBS) and 100 U ml−1 recombinant colon stimulating factor from PeproTech, Rocky Hill, NJ, USA). After 7 days in culture at 37 °C with 5% CO2, the adherent cells (>95% are expected to be macrophages) were harvested for assays38.
Mammalian whole-cell electrophysiology
HEK293T, Cos-1 and CHO cells were used for the heterologous expression experiments and were transfected using Lipofectamine 2000 (Invitrogen). The pipette solution contained 147 mM Cs, 120 mM methane-sulphonate, 4 mM NaCl, 10 mM EGTA, 2 mM Na2-ATP, 2 mM MgCl2 and 20 mM HEPES (pH 7.2; free [Ca2+]i <10 nM). The standard extracellular bath solution (modified Tyrode's solution) contained 153 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 20 mM HEPES and 10 mM glucose (pH 7.4). The 'Low pH Tyrode' solution contained 150 mM Na-gluconate, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES and 10 mM MES (pH 4.6). All bath solutions were applied via a perfusion system to achieve a complete solution exchange within a few seconds. Data were collected using an Axopatch 2A patch clamp amplifier, Digidata 1440 and pClamp 10.0 software (Axon Instruments). Whole-cell currents were digitized at 10 kHz and filtered at 2 kHz. All experiments were conducted at room temperature (21–23 °C), and all recordings were analysed with pClamp 10.0 and Origin 8.0 (OriginLab, Northampton, MA, USA).
Endolysosomal electrophysiology
Endolysosomal electrophysiology was performed in isolated endolysosomes using a modified patch-clamp method11,14. Briefly, cells were treated with 1 μM vacuolin-1 for 2–5 h to increase the size of endosomes and lysosomes. Whole-endolysosome recordings were performed on isolated enlarged LELs. The bath (internal/cytoplasmic) solution contained 140 mM K-gluconate, 4 mM NaCl, 1 mM EGTA, 2 mM Na2-ATP, 2 mM MgCl2, 0.39 mM CaCl2, 0.2 mM GTP and 10 mM HEPES (pH adjusted with KOH to 7.2; free [Ca2+]i ~100 nM based on the Maxchelator software (http://maxchelator.stanford.edu/)). The pipette (luminal) solution consisted of a 'Low pH Tyrode's solution' with 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM MES and 10 mM glucose (pH 4.6).
Fura-2 Ca2+ imaging
Cells were loaded with 5 μM Fura-2 AM in the culture medium at 37 °C for 60 min. Fluorescence was recorded at different excitation wavelengths using an EasyRatioPro system (PTI). Fura-2 ratios (F340/F380) were used to monitor changes in intracellular [Ca2+] upon stimulation. GPN (200–400 μM, a lysosome-disrupting agent) and Baf-A1 (500 nM, a V-ATPase inhibitor) were used as positive controls to induce Ca2+ release from lysosome and acidic stores, respectively39. Ionomycin (1 μM) was added at the conclusion of all experiments to induce a maximal response for comparison. For endogenous lysosomal Ca2+ release measurement, Ca2+ imaging was carried out within 0.5–3 h after plating and when cells still exhibited a round morphology.
GCaMP3 Ca2+ imaging
At 18–24 h after transfection with GCaMP3-ML1, CHO or human fibroblast cells were trypsinized and plated onto glass coverslips. Most experiments were carried out within 0.5–3 h after plating when cells still exhibited a round morphology. The fluorescence intensity at 470 nm (F470) was monitored using the EasyRatioPro system. Lysosomal Ca2+ release was measured under a 'low' external Ca2+ solution, which contained 145 mM NaCl, 5 mM KCl, 3 mM MgCl2, 10 mM glucose, 1 mM EGTA and 20 mM HEPES (pH 7.4). Ca2+ concentration in the nominally free Ca2+ solution is estimated to be 1–10 μM. With 1 mM EGTA, the free Ca2+ concentration is estimated to be <10 nM based on the Maxchelator software (http://maxchelator.stanford.edu/).
LacCer-BODIPY trafficking
Cells for the Lactosylceramide (LacCer)-BODIPY uptake experiment were grown on glass coverslips and incubated with 5 μM bovine serum albumin-complexed LacCer in 'Tyrode' solution for 45 min at room temperature. Cells were then washed twice with PBS and incubated with normal DMEM/F12 medium containing 10% FBS at 37 °C for 1 h. For the correction experiments, NPC1−/− macrophages were pretreated with ML-SA1 (10 μM) for 12 h, and chased in the presence of ML-SA1 (10 μM). Images were taken using a Leica (TCS SP5) confocal microscope.
Filipin staining
Cells were fixed in 3% paraformaldehyde for 1 h, washed twice with PBS and then incubated with 1.5 mg ml−1 glycine in PBS for 10 min. Cells were then stained for 2 h with 0.05 mg ml−1 filipin25 in PBS supplemented with 10% FBS. All procedures were conducted at room temperature. Images were taken using a fluorescence microscope with a UV filter. The lysosome-like storage organelle filipin intensity was calculated based on low- and high-threshold intensities in the image34.
Lysenin staining
Cells were fixed with 4% Paraformaldehyde (PFA) for 20 min, blocked with 1% bovine serum albumin in Phosphate Buffer Saline with 0.1%(v/v) Tween-20 (PBST) for 2 h and then incubated with lysenin (0.1 μg ml−1, Peptides International) for 20 h at 4 °C. The cells were then incubated with anti-lysenin antibody (Peptides International) for 1 h, followed by Alexa Fluor 488 anti-rabbit (Invitrogen) for 45 min. Images were taken using a Leica confocal microscope.
Reagents
Sphingomyelins (Sigma) were dissolved in 'Tyrode' or 'Low pH Tyrode' solutions by sonication. β-cyclodextrin, filipin and fluoxetine were obtained from Sigma; desipramine was purchased from Santa Cruz; C6-ceramide was obtained from Matreya, LLC; sphingomyelinase was obtained from MP Biomedicals; SF-51 and ML-SA1 were purchased from Princeton BioMolecular Research Inc; PI(3,5)P2 was purchased from Echelon; U18666A was purchased from Enzo; LacCer-BODIPY was purchased from Invitrogen, and lysenin and anti-lysenin antibody were purchased from Peptides International.
Data analysis
Data are presented as the mean±s.e.m.. Statistical comparisons were made using analysis of variance. A P-value <0.05 was considered statistically significant.
Additional information
How to cite this article: Shen, D. et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 3:731 doi: 10.1038/ncomms1735 (2012).
References
de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 7, 847–849 (2005).
Parkinson-Lawrence, E. J. et al. Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda) 25, 102–115 (2010).
Schuchman, E. H. Acid sphingomyelinase, cell membranes and human disease: lessons from Niemann-Pick disease. FEBS Lett. 584, 1895–1900 (2010).
Rosenbaum, A. I. & Maxfield, F. R. Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 116, 789–795 (2011).
Vitner, E. B., Platt, F. M. & Futerman, A. H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 285, 20423–20427 (2010).
Puri, V. et al. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol. 1, 386–388 (1999).
Kiselyov, K., Yamaguchi, S., Lyons, C. W. & Muallem, S. Aberrant Ca2+ handling in lysosomal storage disorders. Cell Calcium 47, 103–111 (2010).
Pryor, P. R., Reimann, F., Gribble, F. M. & Luzio, J. P. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 7, 1388–1398 (2006).
Vergarajauregui, S. & Puertollano, R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7, 337–353 (2006).
Thompson, E. G., Schaheen, L., Dang, H. & Fares, H. Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol. 8, 54 (2007).
Dong, X. P. et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996 (2008).
Venkatachalam, K. et al. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 135, 838–851 (2008).
Cheng, X., Shen, D., Samie, M. & Xu, H. Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 584, 2013–2021 (2010).
Dong, X. P. et al. PI(3,5)P(2) controls membrane traffic by direct activation of mucolipin Ca release channels in the endolysosome. Nat. Commun. 1, 38 (2010).
Shen, D., Wang, X. & Xu, H. Pairing phosphoinositides with calcium ions in endolysosomal dynamics: phosphoinositides control the direction and specificity of membrane trafficking by regulating the activity of calcium channels in the endolysosomes. Bioessays 33, 448–457 (2011).
Bargal, R. et al. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 26, 118–123 (2000).
Bassi, M. T. et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 67, 1110–1120 (2000).
Sun, M. et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 9, 2471–2478 (2000).
Grimm, C. et al. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc. Natl Acad. Sci. USA 104, 19583–19588 (2007).
Xu, H., Delling, M., Li, L., Dong, X. & Clapham, D. E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl Acad. Sci. USA 104, 18321–18326 (2007).
Grimm, C. et al. Small molecule activators of TRPML3. Chem. Biol. 17, 135–148 (2010).
Zhang, Y. et al. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc. Natl Acad. Sci. USA 104, 17518–17523 (2007).
Abe, K. & Puertollano, R. Role of TRP channels in the regulation of the endosomal pathway. Physiology (Bethesda) 26, 14–22 (2011).
Tian, L. et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 (2009).
Lloyd-Evans, E. et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 (2008).
Cadigan, K. M., Spillane, D. M. & Chang, T. Y. Isolation and characterization of Chinese hamster ovary cell mutants defective in intracellular low density lipoprotein-cholesterol trafficking. J. Cell Biol. 110, 295–308 (1990).
Cruz, J. C., Sugii, S., Yu, C. & Chang, T. Y. Role of Niemann-Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J. Biol. Chem. 275, 4013–4021 (2000).
Liscum, L. & Faust, J. R. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 264, 11796–11806 (1989).
Lloyd-Evans, E. & Platt, F. M. Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic 11, 419–428 (2010).
Hurwitz, R., Ferlinz, K. & Sandhoff, K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol. Chem. Hoppe Seyler 375, 447–450 (1994).
Kornhuber, J. et al. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J. Med. Chem. 51, 219–237 (2008).
Pagano, R. E. & Chen, C. S. Use of BODIPY-labeled sphingolipids to study membrane traffic along the endocytic pathway. Ann. N Y Acad. Sci. 845, 152–160 (1998).
Kolter, T. & Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 584, 1700–1712 (2010).
Devlin, C. et al. Improvement in lipid and protein trafficking in Niemann-Pick C1 cells by correction of a secondary enzyme defect. Traffic 11, 601–615 (2010).
Chen, C. S., Bach, G. & Pagano, R. E. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc. Natl Acad. Sci. USA 95, 6373–6378 (1998).
Chen, C. S., Patterson, M. C., Wheatley, C. L., O'Brien, J. F. & Pagano, R. E. Broad screening test for sphingolipid-storage diseases. Lancet 354, 901–905 (1999).
Venugopal, B. et al. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. Am. J. Hum. Genet. 81, 1070–1083 (2007).
Link, T. M. et al. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat. Immunol. 11, 232–239 (2010).
Calcraft, P. J. et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600 (2009).
Acknowledgements
This work was supported by NIH RO1 grants (NS062792 to H.X and NS063967 to A.P.L.). WT control and NPC (NPC1−/−) CHO cells were a gift from Dr. T.Y. Chang at Dartmouth Medical School. We are grateful to Drs Susan Slaugenhaupt and Jim Pickel for TRPML1−/− mice, Dr. Loren Looger for the GCaMP3 construct, Dr. Yusuf Hannun for the DsRed-aSMase construct, and Richard Hume and Ken Cadigan for comments on an earlier version of the manuscript. We appreciate the encouragement and helpful comments from other members of the Xu laboratory.
Author information
Authors and Affiliations
Contributions
H.X., D.S., A.P.L. and H.D.S. designed the research; D.S., X.W., X.L., X.Z., Z.Y., S.D., X.-P.D. and T.Y. performed the research; X.L. and D.S. contributed the new regents; D.S., X.W., X.L., X.Z., Z.Y., T.D., A.P.L., H.D.S. and H.X. analysed the data; and H.X. and D.S. wrote the paper with inputs from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S6 (PDF 2420 kb)
Rights and permissions
About this article
Cite this article
Shen, D., Wang, X., Li, X. et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3, 731 (2012). https://doi.org/10.1038/ncomms1735
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms1735
This article is cited by
-
Lysosome blockade induces divergent metabolic programs in macrophages and tumours for cancer immunotherapy
Journal of Experimental & Clinical Cancer Research (2023)
-
Induction of lysosomal exocytosis and biogenesis via TRPML1 activation for the treatment of uranium-induced nephrotoxicity
Nature Communications (2023)
-
Lysosomal Ca2+ as a mediator of palmitate-induced lipotoxicity
Cell Death Discovery (2023)
-
Liver lipophagy ameliorates nonalcoholic steatohepatitis through extracellular lipid secretion
Nature Communications (2023)
-
Iron-induced cytotoxicity mediated by endolysosomal TRPML1 channels is reverted by TFEB
Cell Death & Disease (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
