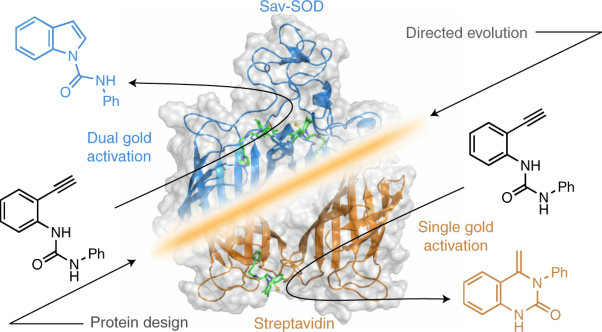
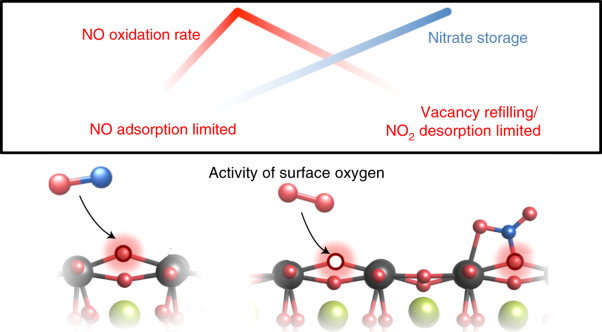
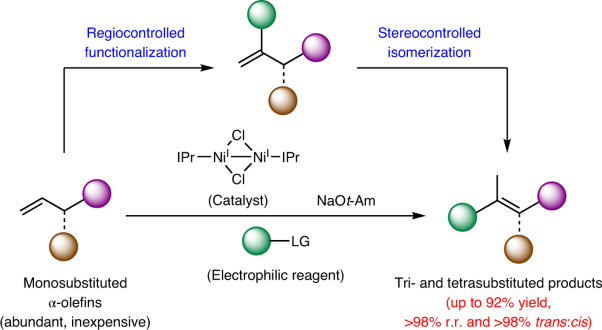
Volume 4 Issue 8, August 2021
Factored in CO2 electroreduction
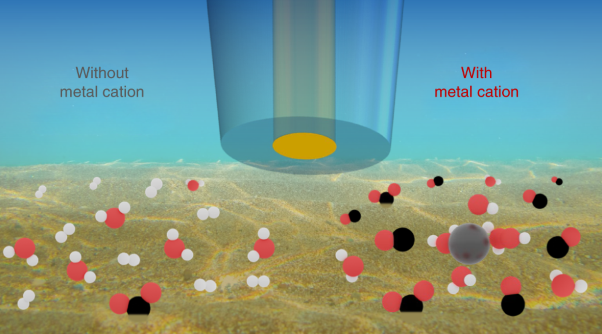
Metal cations are known to influence the performance of the electrochemical reduction of CO2, but their specific role is still unclear. Here, Marc Koper and co-workers investigate the role of alkali cations in CO2 electroreduction onAu, Ag and Cu electrodes with and without metal cations. Based on their results using electrochemical measurements, scanning electrochemical microscopy in the surface-generation tip-collection mode and ab initio molecular dynamics, the authors find that the reaction does not take place without a metal cation.
See Monteiro et al.
Image: Image courtesy of Katrina Goretskaya Cover Design: Marina Spence