Volume 2 Issue 2, February 2019
Shifting reactivity pathways
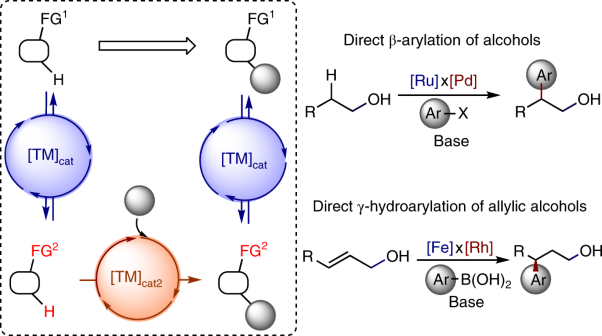
Due to electronic effects, certain activating groups in organic molecules can increase the reactivity of nearby bonds. Now, Lichosyt et al. have shown that such activating groups can be transiently introduced into otherwise unreactive molecules by catalytic reversible reactions. When combined with subsequent catalytic functionalization reactions, the constructed networks of reactions enable the simple functionalization of normally unreactive sites.
See Lichosyt et al.
Image: Adrian Grosu (stock.adobe.com) and Paweł Dydio (University of Strasbourg). Cover Design: Karen Moore.

Editorial
-
Advertisement