
Abstract
Epidemiological and experimental observations suggest that chronic microbial colonization can impact the immune control of other unrelated pathogens contracted in a concomitant or sequential manner. Possible interactions between Mycobacterium tuberculosis infection and persistence of other bacteria have scarcely been investigated. Here we demonstrated that natural colonization of the digestive tract with Helicobacter hepaticus in mice is concomitant with modification of the gut microbiota, subclinical inflammation, and drastic impairment of immune control of the growth of subsequently administered M. tuberculosis, which results in severe lung tissue injury. Our results provided insights upon the fact that this prior H. hepaticus colonization leads to failures in the mechanisms that could prevent the otherwise balanced cross-talk between M. tuberculosis and the immune system. Such disequilibrium ultimately leads to the inhibition of control of mycobacterial growth, outbreak of inflammation, and lung pathology. Among the dysregulated immune signatures, we noticed a correlation between the detrimental lung injury and the accumulation of activated T-lymphocytes. Our findings suggest that the impact of prior Helicobacter spp. colonization and subsequent M. tuberculosis parasitism might be greater than previously thought, which is a key point given that both species are among the most frequent invasive bacteria in human populations.
Similar content being viewed by others

Introduction
Consequences of multiple microbial colonization of the host are poorly understood. However, numerous epidemiological and experimental observations show that acute or chronic infections, and consequent host immune responses, can impact immunity, and thereby sensitivity to either concomitantly or sequentially contracted unrelated pathogens.1, 2 Investigation of the impact of symptomatic or asymptomatic microbial colonization on other pulmonary diseases, notably tuberculosis, can be of utmost importance, particularly in endemic tropical areas with a high risk of infection with unrelated pathogens. With one-third of the world’s population latently parasitized with Mycobacterium tuberculosis, ≈8 million new cases of active tuberculosis, and more than 1.4 million deaths per year, tuberculosis remains a leading cause of morbidity and mortality worldwide (http://www.who.int/tb/country/en/).
The effects of concurrent infection with human immunodeficiency virus and the resultant immune depression regarding the control of M. tuberculosis are well documented.3 Deleterious tuberculosis symptoms have also been largely described as a consequence of pre-existing intestinal helminth infections, setting up a T-helper type 2-biased environment that leads to the accumulation of alternatively activated macrophages and decreases in the protective antimycobacterial immunity.4, 5, 6, 7 Consistently, deleterious effects of plasmodium infection have been reported on the microbiocidal functions of macrophages and dendritic cells as well as on alterations of the cytokine balance, which impairs protective immunity to mycobacteria.8, 9 It is also known that repeated administration of attenuated Mycobacterium bovis BCG (Bacillus Calmette-Guérin), as well as repeated exposure of M. tuberculosis-infected animals to live mycobacteria or mycobacterial antigens, may induce immune-mediated symptoms, such as exacerbated inflammation and lung tissue injury, known as the Koch phenomenon, which is linked to the overstimulation of the interleukin-23/-17 (IL-23/IL-17) axis and granulocytic inflammation.10 Moreover, intranasal treatment of M. tuberculosis-colonized mice by agents that stimulate prolonged inflammation and notably sustained type I interferons (IFNs) leads to exacerbated tuberculosis symptoms.11 Nevertheless, the possible impacts of unrelated chronic bacterial colonization on M. tuberculosis-caused pathogenesis have been barely investigated.
Helicobacter hepaticus is an enterohepatic bacterium that can be found in the mucosal layer of the gastrointestinal tract or in liver tissue. In humans, H. hepaticus can be associated with diseases of the liver and biliary tracts.12 In susceptible strains of mice, H. hepaticus can cause chronic active hepatitis, liver cancer, and inflammatory bowel diseases13 and remains the most prevalent Helicobacter in laboratory mouse colonies worldwide.14, 15 Here, we report a concomitance between the natural colonization of the digestive tract by H. hepaticus, modified gut microbiota, and the drastic impairment of the immune control of the growth of subsequently administered M. tuberculosis, which results in exacerbated lung tissue injury and intensified tuberculosis-associated immunopathology. This final outcome is associated with an outbreak of inflammation, uncontrolled innate immunity, and dramatic accumulation of activated T cells in the lungs. Our observations suggest that far from the situation that occurs in specific pathogen-free laboratory mice with a resting immune system, highly probable cocolonization with unrelated bacteria in nature, notably in the endemic area of tuberculosis, can considerably impact M. tuberculosis-induced human lung pathogenesis.
Results
Gavage of B10.D2 mice with H. hepaticus results in a decreased control of M. tuberculosis growth
To investigate the possible interference between a chronic bacterial infection and the development of active tuberculosis, we first evaluated the impact of experimentally induced H. hepaticus colonization on the growth of subsequently delivered M. tuberculosis. B10.D2 mice were infected by gastric gavage with the natural enterohepatic mouse pathogen H. hepaticus 3B1 strain.16 At 2 months post gavage, the fecal matter DNA was positive for H. hepaticus, as monitored by Helicobacter-specific PCR (Figure 1a), followed by sequencing showing the establishment of a chronic H. hepaticus colonization. These mice were challenged by aerosol route with M. tuberculosis H37Rv strain at 3 months post gavage. At 2 months post challenge, statistically higher mycobacterial loads were notably detected in the lungs of these H. hepaticus-colonized mice, compared with the controls (Figure 1b). Thus, colonization with H. hepaticus seems to interfere with the control of the growth of M. tuberculosis.

Impact of experimental helicobacter chronic infection on the control of Mycobacterium tuberculosis growth. (a) Experimental chronic infection of the digestive tract by Helicobacter hepaticus, as determined by helicobacter-specific PCR on the fecal matter DNA of B10.D2 mice at 2 months after gavage with H. hepaticus 3B1 strain. (b) Control mice gavaged with peptone (Pept.) broth (n=6) or H. hepaticus 3B1 strain (n=10) were challenged with M. tuberculosis, and mycobacterial loads were determined at 8 weeks post challenge. *P<0.05, NS, nonsignificantly different, as determined by Student’s t-test and Mann–Whitney tests. Error bars represent mean±s.d. The results are representative of at least two independent experiments. CFU, colony-forming unit.
Characterization of naturally H. hepaticus-colonized B10.D2 mice and their gut microbiota
We further investigated a naturally H. hepaticus-colonized B10.D2 mouse breeding. An exhaustive sanitary control for mouse pathogens, including endoparasites, ectoparasites, bacteria, and viruses, known as murine pathogens, only revealed the presence of Helicobacter spp. in this colony (Supplementary Table S1 online). A PCR amplification with primers specific for Helicobacter 16S rRNA performed on the fecal DNA revealed colonization of the digestive tract by Helicobacter. Sequencing of the PCR products identified only the presence of H. hepaticus. For further comparative studies vs. appropriate controls, we rederived B10.D2 mice, by means of embryo transfer, from this naturally H. hepaticus-colonized breeding.
Comparative histopathological analyses of B10.D2 mice showed a moderate subclinical hepatic inflammation in naturally H. hepaticus-colonized individuals (Supplementary Figure S1A). Higher percentages of CD11b+ Gr1+ innate immune cells in the spleen (Supplementary Figure S1B) and of neutrophils, i.e., Ly6C− Ly6G+ within the CD11b+ Gr1+ subset, were detected in the spleen and lungs (Supplementary Figure S1B,C) of the naturally H. hepaticus-colonized B10.D2 mice.
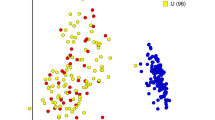
Beyond the analysis of mouse-specific pathogens, we compared the gut microbiota of the naturally H. hepaticus-colonized mice to the Helicobacter-free rederived B10.D2 controls by high-throughput 16S rRNA gene sequencing. On average, 7,209±2,278 high-quality reads were produced from each fecal DNA sample extracted from individual mice of each group (n=6) and reads were grouped in a total of 4,526±1,560 operational taxonomic units. The α-diversity plots showed an overall lower biodiversity (P value of the permutation-based t-test: 0.002) in the naturally H. hepaticus-colonized mice, compared with the controls (Figure 2a), which is consistent with previous reports.17 Moreover, the principal coordinate analysis (PcoA) plots derived from β-diversity (for both weighted and unweighted Unifrac distance) showed a clear significant separation (P values of the Adonis test: 0.005) (Figure 2b), and was also confirmed by analyzing the relative abundance of bacterial groups (Figure 2c). Naturally H. hepaticus-colonized mice were found to have a gut microbiota enriched in Bacteroidaceae and other unclassified bacteroidales (average relative abundance as compared with noninfected mice: +11.3% and +5.8%, respectively), and were associated with a strong reduction in Clostridiales, Ruminococcaceae, Lachnospiraceae, and Prevotellaceae (average relative abundance as compared with noninfected mice: −5.3%, −1.8%, −2.5%, and −4.8%, respectively) (Supplementary Table S2). As expected, the Helicobacteriaceae family and Helicobacter genus were found only in the naturally H. hepaticus-colonized mice.

Comparative study of the gut microbiota composition in B10.D2 mice, specific pathogen-free or naturally colonized with Helicobacter hepaticus. (a) α-Diversity: H. hepaticus colonization is characterized by a reduced biodiversity in the gut microbiota. Rarefaction plots of chao1 index values for naturally H. hepaticus-colonized (red) and specific pathogen-free (blue) mice show a statistically significant difference between the two curves (P=0.002, as determined by permutation-based nonparametric t-test). At the same time, having reached a plateau, the curves suggest that the number of sequencing reads obtained was sufficient to completely describe the gut microbiota biodiversity. (b) β-Diversity: Individual mouse microbiota clearly separate on the basis of colonization by H. hepaticus. Principal coordinate analysis (PCoA) from weighted Unifrac distances among samples reveals that H. hepaticus-sustained colonization is the main factor discriminating the samples, as expressed by the relative contribution of PCoA component 1 (accounting for 47.53% of the total variance). Each sample collected from naturally H. hepaticus-colonized mice (red) displayed negative values of PCoA_1, whereas samples collected from H. hepaticus-free (blue) mice displayed positive PCoA-1 values. Differences in distance values are statistically significant (P=0.002). (c) Hierarchical clustering: Analysis of the relative abundance of bacterial families reveals a different microbiota composition between H. hepaticus-positive or -negative mice. Hierarchical clustering, created using Spearman’s correlation as metric and Ward distance as linkage, shows a clear separation between B10.D2 mice H. hepaticus-colonized (red) or control (blue). Each sample is represented by the relative abundance of the most present bacterial families. All bacterial groups with relative abundance <1% on average were grouped under the “Other” label.
Severe M. tuberculosis-caused pathogenesis in naturally H. hepaticus-colonized mice
We compared the sensitivity to M. tuberculosis-caused pathogenesis of the naturally H. hepaticus-colonized B10.D2 mice, the B10.D2 controls recently imported from the Jackson Laboratories (Bar Harbor, ME) or their Helicobacter-free rederived counterparts (Figure 3a). At 2 months after M. tuberculosis challenge by aerosol route, the mice harboring the two pathogens exhibited moribund features including distress, prostration, piloerection, and arched back and displayed 3 or 2 log10 more colony-forming unit of mycobacteria, in different organs, as compared with the controls. The sustained natural colonization of B10.D2 mice by H. hepaticus and the modified microbiota were also concomitant with deleterious lung tissue injury (Figure 3b) and increased percentages of the lesional surface lung parenchyma (Figure 3c), as evaluated at 8 weeks after M. tuberculosis challenge (Figure 3b,c). The H. hepaticus-colonized, M. tuberculosis-infected mice showed substantial infiltration by macrophages, epitheloid cells, lymphocytes/plasma cells, and neutrophils, as well as marked granulomatous caseous necrosis and the presence of numerous acid-fast bacilli (Figure 3d).

Severe Mycobacterium tuberculosis-caused lung tissue injury in Helicobacter hepaticus-colonized mice. (a) B10.D2 mice, naturally colonized with H. hepaticus, rederived from the H. hepaticus-positive colony, or recently imported from the Jackson Laboratories (control (Ctrl)) (n=6 per group), were given M. tuberculosis through aerosol. The input of M. tuberculosis in the lungs was between 150 and 200 colony-forming unit per mouse, as determined at day 1 post challenge, whether in “clean” or naturally H. hepaticus-colonized mice. The mycobacterial loads were determined at 8 weeks post challenge in individual mice. NS=nonsignificantly different. *, **Statistically significant, as determined by one-way analysis of variance (ANOVA) test with Tukey’s correction, P<0.05, P<0.005, respectively. The results are representative of eight independent experiments. (b) Tuberculous lesions in the lungs of controls or naturally H. hepaticus-colonized B10.D2 mice at week 7 after M. tuberculosis delivery, as detailed in (a). Bar=750 μm. (c) Increased percentage of the lesional surface of lung parenchyma in H. hepaticus-colonized mice compared with the control individuals, as determined by Student’s t-test and Mann–Whitney tests, *P<0.05. (d) Lesions in the lungs of B10.D2 mice at week 7 post infection (p.i.), as shown after hematoxylin and eosin (H&E) staining and illustrated at higher magnification (upper panels and left lower panel) and after Ziehl–Neelsen staining (right lower panel), revealing the presence of numerous acid-fast bacilli. Bars=upper panels, 500 μm; lower panel left, 100 μm; and lower panel right, 10 μm. The results are representative of at least three experiments with n=3 per group.
We also compared these naturally H. hepaticus-colonized B10.D2 mice to several other H. hepaticus-free laboratory mouse strains from our breeding and housed in the same animal facilities, including B6, B10, B10.S, and SJL mice (Figure 4a). At 10 days after M. tuberculosis challenge, the mycobacterial loads were comparable in all these experimental groups showing that at preadaptive stages the growth of mycobacteria is not impacted by Helicobacter colonization. At 5 weeks post challenge, only the naturally H. hepaticus-colonized B10.D2 mice exhibited morbidity and had significantly higher mycobacterial loads. Moreover, at 7 weeks post challenge, 3 or 2 log10 more colony-forming unit of mycobacteria were again detected in the organs of these B10.D2 mice. We also detected significantly higher lung mycobacterial loads at 5 weeks post challenge in the organs of Mus spretus SEG wild-type mice (Figure 4b), which were naturally colonized with H. hepaticus (Figure 4c). Therefore, the enhanced susceptibility to M. tuberculosis-mediated disease subsequent to H. hepaticus colonization is not restricted to the B10.D2 mouse strain.

Mycobacterium tuberculosis burden in the lungs, spleen, and liver of B10.D2 mice according to their Helicobacter hepaticus-positive or -negative status. (a) B6, B10, B10.S, and SJL control mice (n=6 per group) and naturally H. hepaticus-colonized B10.D2 mice were given M. tuberculosis through aerosol. Mycobacterial loads were determined at 10 days, 5 or 7 weeks post challenge in different organs of individual mice. ND=not determined. The results are representative of at least 7–8 independent experiments. (b) B6 or wild M. spretus SEG mice (n=4 per group) with natural H. hepaticus colonization were given M. tuberculosis as described in (a). Mycobacterial loads were determined at 4 weeks post challenge in individual mice. NS=nonsignificantly different. *, ** or ****Statistically significant, as determined by one-way analysis of variance (ANOVA) with Tukey’s correction, P<0.05, P<0.005 or P<0.0001, respectively. The results are representative of two experiments. (c) Detection of Helicobacter-specific antibodies in the wild-type M. spretus SEG mice, as determined by ELISA (enzyme-linked immunosorbent assay) before M. tuberculosis challenge. CFU, colony-forming unit.
Early postnatal (<72 h after birth) transfer of B10.D2 pups born from naturally H. hepaticus-colonized mothers to Helicobacter-free B10.D2 foster mothers resulted in the eradication of this colonization (Supplementary Figure S2A). Consistently, the adopted mice did not display uncontrolled lung mycobacterial load subsequent to M. tuberculosis challenge (Supplementary Figure S2B). Therefore, in numerous experimental frameworks, the natural H. hepaticus colonization coincides with a severe M. tuberculosis-mediated lung pathogenesis.
Extensive inflammation in the lungs of the H. hepaticus-colonized, M. tuberculosis-infected mice
Before the onset of adaptive immunity, i.e., at 10 days after M. tuberculosis challenge, the total numbers of leukocytes in the lung and spleen of the naturally H. hepaticus-colonized or H. hepaticus-free B10.D2 mice were comparable (Figure 5a). At this time point, the percentages and absolute numbers of CD11bhi or CD11chi cells and the percentages of F4/80+ cells within the CD11bhi cells were also comparable in these two groups (Figure 5b). At 5 or 7 weeks after M. tuberculosis challenge, marked leukocyte accumulation in the lungs and splenomegalia were detected in the naturally H. hepaticus-colonized mice (Figure 5a). In the lungs of the latter, at these time points, we detected: (i) increased percentages and absolute numbers of CD11bhi cell population, (ii) decreased percentages, yet increased numbers of CD11chi dendritic cells, and (iii) comparable proportions, yet increased numbers, of F4/80+ mature macrophages inside the CD11bhi subset (Figure 5b,c). Before the challenge (Supplementary Figure S1C) and at any time point post challenge (Figure 5b,c), higher percentages and the number of neutrophils (Ly6G+) were detected in the lungs of the naturally H. hepaticus-colonized mice.

Immune cell composition in the organs of coinfected mice. (a) Total numbers of leukocytes, recovered on Optiprep gradient from lung homogenates or from the spleen homogenates in untreated (day 0) or Mycobacterium tuberculosis-delivered controls or naturally Helicobacter hepaticus-infected mice at day 10, week 5 or 7 postinfection (p.i.). *, **P<0.05 or P<0.005, respectively, as determined by Student’s t-test and Mann–Whitney tests. (b) Percentages and total numbers of different innate immune cell subsets in the lungs of M. tuberculosis-delivered controls or naturally H. hepaticus-infected mice, at 10 days, 5 or 7 weeks post challenge, as determined by cell counting and flow cytometric analyses. Percentages of F4/80+ or Ly6G+ cells are calculated compared with the total CD11bhi cells. (c) Representative profile of the innate immune cell subsets in the lungs of infected mice, at week 7 p.i., as determined by flow cytometry. Comparative study of the expression level (d) and mean fluorescence intensities (MFIs) (e) of CD11b, CD103, and CD205 markers on the CD11chi dendritic cells recovered from the lungs, at week 7 p.i. ***, ****Statistically significant, as determined by one-way analysis of variance (ANOVA) with Tukey’s correction, P<0.001 and P<0.0001, respectively. The results are representative of two experiments with n=3 per group.
No differences were observed in the expression levels of CD11b, CD103, and CD205 by the lung CD11chi dendritic cells in the rederived or H. hepaticus-colonized mice before the M. tuberculosis challenge (Figure 5d,e). In contrast, at 7 weeks after the challenge, the lung CD11chi dendritic cells of the H. hepaticus-colonized mice displayed increased levels of CD11b, CD103, and CD205, a phenotype that seems to be linked to their more mature and more migratory characteristics, and was not observed in the lung dendritic cells of the rederived Helicobacter-free counterparts. At adulthood, the innate cell compartments of B10.D2 mice, neonatally transferred to clean foster mothers, and which eradicated the Helicobacter colonization, were comparable to that of rederived B10.D2 mice (data not shown).
To further characterize the immuno(patho)logical features of these mice, we then analyzed the inflammatory cytokine and chemokine signature in total lung homogenates of the Helicobacter-free mice or naturally H. hepaticus-colonized individuals, either before or 7 weeks after an M. tuberculosis challenge (Figure 6a). We did not detect significant differences in the cytokine production profiles before M. tuberculosis challenge. Nevertheless, hypercytokinemia, notably concerning tumor necrosis factor-α, IFN-γ, IL-1α, IL-1β, IL-6, and IL-17, and substantially higher amounts of CCL2, CCL3, CCL4, CCL5, CXCL1, CXCL2, CXCL9, and CXCL10 chemokines were found in the mice harboring the two pathogens compared with the Helicobacter-free, M. tuberculosis-challenged counterparts. These excessive inflammatory responses were most seemingly the consequence of a sustained overstimulation of the innate immune cells, which were not able to control M. tuberculosis growth despite their high intensity. The weak amounts of IL-5 detected in M. tuberculosis-delivered mice were similar in both groups and no IL-4 was detected, which showed that the failure in the control of M. tuberculosis growth was not due to a switch to a nonprotective Th2-biased environment.

Profile of lung innate immune signature and T-cell activation status in Helicobacter-free or the naturally Helicobacter hepaticus-colonized mice before and after Mycobacterium tuberculosis delivery. (a) Amounts of cytokines and chemokines in the lung homogenates of Helicobacter-free rederived or naturally H. hepaticus-colonized B10.D2 mice, before or 7 weeks after aerosol challenge with M. tuberculosis (n=3 per group). Levels of cytokines and chemokines were determined by multiple Milliplex Kit Assay (Millipore, Guyancourt, France) and a Luminex X-100 Reader (Hertogenbosch, The Netherlands), whereas interferon-α (IFN-α) was quantified by ELISA (enzyme-linked immunosorbent assay). *, **Statistically significant, as determined by Student’s t-test and Mann–Whitney tests, P<0.05, P<0.005, respectively. (b) Percentages (mean±s.d.) (n=3 per group) and (c) representative cytometric profiles of different populations as a function of expression of activation markers by CD4+ or CD8+ T cells in mice, rederived Helicobacter-free or naturally colonized with H. hepaticus, before or at different time points after M. tuberculosis aerosol challenge. The data are representative of two independent experiments with n=3 per group. *Statistically significant, as determined by Student’s t-test and Mann–Whitney tests, P<0.05. CCL5, chemokine (C–C motif) ligand 5; CXCL5, C–X–C motif chemokine 5; IFN, interferon; IL, interleukin; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.
Accumulation of activated T cells in the lungs of the H. hepaticus-colonized, M. tuberculosis-infected mice
At 3 or 7 weeks post challenge, the mice harboring both pathogens and the M. tuberculosis-delivered rederived controls displayed comparable IFN-γ T-splenocyte responses against purified protein derivative of tuberculin or specific to the protective TB10.4 or Ag85A mycobacterial immunogens (Supplementary Figure S3A). Thus, the development of M. tuberculosis-specific Th1 responses in the secondary lymphoid organs did not seem to be inhibited at the early (3 weeks post challenge) and later (7 weeks post challenge) time points of the postadaptive stage of M. tuberculosis infection.
Before M. tuberculosis challenge, compared with the controls, naturally H. hepaticus-colonized mice had similar lung CD4+ and CD8+ T-cell activation profiles, as indicated by the surface expression of CD27 or CD44 vs. CD45RB (Figure 6b,c). At 10 days post M. tuberculosis challenge, no modifications of the T-cell compartments were detected in these groups (Figure 6b). At 5 weeks post M. tuberculosis challenge, higher percentages of CD27− CD45RB− were detected within the lung CD4+ T-cell subset of H. hepaticus-colonized mice. The majority of the lung T cells characteristically upregulated CD44 in the M. tuberculosis-challenged controls, whereas almost all of the CD4+ and CD8+ T cells in the mice harboring the two pathogens reverted to a CD44− phenotype (Figure 6b,c), seemingly as a consequence of substantial and sustained activation and ultimate differentiation. At this time point after the M. tuberculosis challenge, the lung CD4+ CD45RB− CD44low CD27low population contained up to 35±2% of KLRG1+ senescent cells in the H. hepaticus-colonized mice vs. 3±1% in their rederived counterparts (Supplementary Figure S3B). Such senescent T cells are admitted to be nonprotector and predictor of active tuberculosis.18
In summary, a pre-existing Helicobacter colonization coincides with a modification of the gut microbiota and a subclinical inflammation, which are correlated, in M. tuberculosis-delivered mice, with an overwhelming stimulation of innate immunity, accumulation of activated T cells, containing substantial proportions of senescent cells, and deleterious lung tissue injury.
Discussion
We report here a drastic defect in the control of M. tuberculosis growth, and an aggravation of M. tuberculosis-induced pathogenesis, in mice that acquired an unrelated natural, nonpulmonary bacterial colonization and that displayed a modified gut microbiota. In fact, a low-dose M. tuberculosis challenge in mice with chronic H. hepaticus colonization resulted in: (i) a marked impairment of the control of mycobacterial growth; (ii) an overwhelming stimulation of the innate immune system in the lungs, as evidenced by hypercytokinemia and a strong increase in chemokine production; (iii) a drastic accumulation of activated lung T cells; (iv) marked lung tissue destruction; (v) caseous granuloma; and (vi) morbidity/mortality. The mycobacterial loads in the lungs and spleen of H. hepaticus-colonized and -uncolonized control mice were comparable at 10 days after M. tuberculosis challenge, i.e., before the onset of adaptive immunity. This observation suggests that, in H. hepaticus-colonized mice, the early first-line antimycobacterial innate immune effectors, such as bacterial uptake or T-cell-independent intracellular antibacterial defense, should not be affected. In H. hepaticus-colonized mice, the causal mechanism(s) of the high susceptibility to develop symptoms of tuberculosis should rather rely on defects in some adaptive immune effectors or components of the innate immunity, which directly support the onset of the adaptive protective immunity. We demonstrated that the aggravation of M. tuberculosis-caused pathogenesis did not result from the absence of mycobacterial antigen presentation, T-cell triggering, or a generalized impairment of adaptive immunity. However, we detected in the mice bearing the two pathogens a notable accumulation of activated pulmonary CD4+ and CD8+ T cells. The mechanisms involved can rely on the extradigestive/systemic dysregulation of the innate immune effectors in Helicobacter-infected mice, such as serum proinflammatory mediators, that might subclinically affect the maintenance of homeostasis in mucosal tissues and local immunity. The exposure of such disturbed immune cells to substantial numbers of various mycobacterial pathogen-associated molecular patterns would then lead to an excessive inflammatory response. Expression of very high amounts of cytokines and chemokines in the lungs of the mice bearing the two pathogens is likely to have a major role in the switch from immune protection to immune pathology.
We detected increased percentages and absolute numbers of neutrophils in the spleen and the lungs of H. hepaticus-colonized B10.D2 mice, before and also at any time points after M. tuberculosis challenge. The role of neutrophils in the control of mycobacterial infections remains largely controversial, and both neutrophil-related enhancement and inhibition of T-cell responses have been reported.19, 20, 21, 22, 23, 24, 25 We have previously demonstrated that depletion of neutrophils by the anti-Ly6G (1A8) monoclonal antibody injection results: (i) in an increase of lung mycobacterial loads during acute mycobacterial infection, but (ii) in a decrease in the mycobacterial loads during the chronic mycobacterial infection.25 Because of the replenishment of neutrophils as soon as 3 days after the last 1A8 administration, a monoclonal antibody-mediated neutrophil depletion will necessitate in the present model of two successive chronic bacterial infections, three intravenously injections per week of 200 μg of Ig per mouse per injection during at least 1 month, encompassing time points before and after M. tuberculosis challenge, which is technically and ethically complex. Future work with mice genetically deficient or conditional mutants for key antimycobacterial immune effectors bearing natural H. hepaticus infection should open avenues to unravel the certainly complex and multifaceted mechanisms leading to such a substantial susceptibility to tuberculosis.
We evidenced a net shift in the gut microbiota in H. hepaticus-infected mice to a proinflammatory condition, characterized by an increase of bacteria belonging to Bacteroidetes phylum and a decrease of bacteria belonging to Firmicutes, which is consistent with previous reports.26 Helicobacter-mediated dysregulation of innate immunity and the concomitant modification of the composition of gut-resident microbiota, the parallel subclinical chronic inflammation and increased neutrophil numbers, correlated with the disequilibration of the host–pathogen balance in favor of M. tuberculosis, ultimately resulting in the inhibition of the immune control of mycobacterial growth, outbreak of inflammation, and detrimental lung tissue destruction.
In human, the lung microbiota of cured patients and healthy individuals is more diverse than that of patients with recurrent tuberculosis or treatment failure cases.27 However, it is not clear whether this increased diversity is one of the causes or a consequence of the disease. In mice, aerosol M. tuberculosis challenge itself leads to the loss of diversity in mouse gut microbiota, as rapidly as a week post challenge.28 The minimum diversity in gut microbiota coincides with the time when the adaptive immunity stabilizes the mycobacterial loads, suggesting the existence of an interplay between the delicate balance of gut microbiota and the distal pulmonary immunity.
H. hepaticus colonizes the lower intestinal tract, causes typhlocolitis, and can induce inflammatory bowel diseases.13 Thus, our observations with H. hepaticus cannot be directly extrapolated to colonization with H. pylori, which occupies the gastric niche. It has been reported that in M. tuberculosis-infected humans, H. pylori colonization may impact IFN-γ T-cell responses to mycobacterial antigens.29 Whether an epidemiological link exists between the most prevalent bacterial pathogens, M. tuberculosis and H. pylori, in nonhuman primates and/or in humans remains to be determined.29, 30, 31 Despite the absence of proof of a causal relationship between H. pylori and respiratory disease, epidemiological associations have been reported between the increased seroprevalence of H. pylori infections and several extradigestive pathologies, including respiratory diseases and pulmonary tuberculosis.32 H. pylori infection in neonatal mice has also been shown to prevent the induction of lung allergic responses.33
In human tuberculosis, in addition to coinfections with human immunodeficiency virus and parasites, acute or chronic infections with unrelated bacteria and environmental microbiota, notably in endemic areas, could be a major factor impacting the degree of M. tuberculosis-induced lung disease. From our observations in naturally H. hepaticus-colonized mice, which displays a net shift in their gastrointestinal microbiota, we may learn more about the potential mechanisms involved in the increased susceptibility to lung parenchymal damage. Our results also demonstrate the need for coordinated epidemiological investigations with a particular emphasis on recording the presence of a potential H. pylori infection in tuberculosis patient cohorts and household contacts. If a correlation between tuberculosis disease development and Helicobacter infection could be established, it would be a major breakthrough for the prediction of M. tuberculosis-associated disease.
Methods
Mice. C57BL/10 (B10), B10.S, and SJL mouse strains were bred at Institut Pasteur (Paris, France) in a dedicated separate room of our animal facility. C57BL/6 (B6) were from Janvier (Le Genest-Saint-Isle, France). B10.D2 (Hc1 H-2d H2-T18c/nSnJ), with 16 backcrossed generations to the B6 background, were originally from Jackson Laboratories, and were bred for more than 16 years in Institut Pasteur. In some experiments, B10.D2 mice recently imported from Jackson Laboratories were also used. SEG wild-type mice were bred at Institut Pasteur. All of the animal studies were performed in agreement with guidelines of the European and French guidelines (Directive 86/609/CEE and Decree 87-848 of 19 October 1987), after approval by the Institut Pasteur Safety, Animal Care and Use Committee and under local ethical committee protocol agreement no. CETEA 2014-0024 and CETEA 00317.02.
Gavages with Helicobacter spp. The H. hepaticus 3B1 (ATCC511449) strain, which harbors the intact H. hepaticus genomic pathogenicity island 1,34 was a kind gift from Dr Josenhans (Hannover Medical School, Hannover, Germany). H. hepaticus (3B1 strain) was grown on blood agar base 2 (Oxoid, Lyon, France) plates supplemented with 10% defibrinated horse blood (bioMérieux, Marcy l’Etoile, France) under microaerophilic conditions at 37 °C. Bacteria were resuspended in peptone broth for the inoculation of mice. Mice were given three successive gavages with ≈1 × 108 colony-forming unit per mouse of the H. hepaticus resuspended in 0.2 ml of peptone water, given every other day.35
M. tuberculosis challenge. Mice were challenged via the aerosol route with the virulent M. tuberculosis strain H37Rv as detailed elsewhere.36 Naturally H. hepaticus-colonized B10.D2 mice, challenged with M. tuberculosis, were killed at 6–8 weeks post mycobacterial challenge, when they showed severe clinical signs of morbidity. For histological analyses, the left lobe of the lungs was fixed in 10% neutral-buffered formalin and embedded in paraffin. Five micrometer sections were cut and stained with hematoxylin and eosin and Ziehl–Neelsen stain.
Flow cytometry. Lungs were first perfused via the right ventricle of the hearth with 5 ml of RPMI and mediastinal lymph nodes were discarded. Low-density cells or adaptive lung immune cells were, respectively, prepared on Optiprep or Ficoll gradients, as detailed previously.36 Cells were first incubated with an anti-CD16/CD32 (clone 2.4G2) monoclonal antibody, followed by cocktails of the indicated monoclonal antibodies, all of which were purchased from BD Pharmingen (Le Pont-de-Claix, France). The stained cells were fixed in 4% paraformaldehyde overnight at 4 °C and were acquired with a CyAn system using the Summit software (Beckman Coulter, Villepinte, France). Data were analyzed with the FlowJo software (Treestar, OR).
Detection of H. hepaticus. DNA was extracted from fecal pellets from individual mice by using the Qiagen DNA Stool Kit in the QiaCube System (Qiagen, Valencia, CA). A duplex PCR assay was used to detect chronic colonization of the digestive tract by Helicobacter spp. in parallel to Lactobacillus as a positive control. The genus-specific primers, 5′-AGCAGTAGGGAATCTTCCA-3′ and 5′-ATT(C or T)CACCGCTACACATG-3′, yielded a 340-bp PCR product of the Lactobacillus 16S rRNA gene as a positive control. Primers 5′-GCTATGACGGGTATCC-3′ and 5′-ACTTCACCCCAGTCGCTG-3′ yielded in a 1,220-bp PCR product of the 16S rRNA gene of all known Helicobacter spp.37 The Helicobacter-specific PCR products were then pyrosequenced38 to identify the Helicobacter spp., based on the 16S DNA sequences (Supplementary Table S3). The sanitary status of the naturally H. hepaticus-colonized B10.D2 mice was determined extensively by Charles Rivers (http://www.criver.com/files/pdfs/rms/rabbit_hmsummary.aspx).
Fecal pellet 16S rRNA gene sequencing and processing. DNA recovery, after extraction from fecal pellets from individual mice, was evaluated using NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE) and 2200 TapeStation Instrument (Santa Clara, CA). For each sample, the V3–V4 region of the 16S rRNA gene was PCR amplified in 25 μl volumes containing 12.5 ng of microbial DNA, 2 × KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Resnova, Rome, Italy), and 200 nM of S-D-Bact-0341-b-S-17/S-D-Bact-0785-a-A-21 primers39 carrying Illumina overhang adapter sequences. Thermal cycle consisted of an initial denaturation at 98 °C for 3 min, 25 cycles of denaturation (95 °C for 30 s), annealing (55 °C for 30 s), and extension (72 °C for 30 s), plus a final extension step at 72 °C for 5 min. Amplicons of about 460 bp were purified with a magnetic bead-based clean-up system (Agencourt AMPure XP; Beckman Coulter, Brea, CA) and sequenced on Illumina MiSeq platform using a 2 × 250 bp paired-end protocol, according to the manufacturer’s instructions (Illumina, San Diego, CA). Briefly, indexed libraries were prepared by limited-cycle PCR using Nextera Technology (Illumina) and cleaned up with AMPure XP magnetic beads (Beckman Coulter). Libraries were pooled at equimolar concentrations, denatured, and diluted to 6 pM before loading onto the MiSeq flow cell. Sequencing reads are available in NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra) under ID PRJNA320730.
Bioinformatics and statistics. Raw sequencing reads were processed as follows: read pairs were firstly merged together by means of PandaSeq assembler (“PAired-eND Assembler for DNA sequences”),40 which performs a local assembly between two overlapping pairs, thus creating a single fragment. Nonoverlapping sequences, fragments shorter than 250 bases, or longer than 900 bases were discarded. Then, sequences were quality filtered using the “split_libraries_fastq.py” utility of the QIIME suite,41 which discards from further analyses any sequence having more than 25% low-quality nucleotides, i.e., having a phred score of 3 or less. Quality-filtered reads were, then, analyzed with the standard QIIME pipeline. Sequences were grouped into operational taxonomic units by clustering together reads at 97% identity or higher using UCLUST42 and taxonomically classified against the Greengenes bacterial 16S rRNA database (release 13_8, http://greengenes.lbl.gov) by using the RDP (Ribosomal Database Project) classifier43 at 50% confidence. Singleton sequences were discarded as possible artifacts. Alpha diversities were calculated according to different microbial diversity metrics, i.e., chao 1, Shannon index, observed species and Faith’s phylogenetic distance, and evaluated to determine whether the chosen subset was representative of the overall microbial diversity within each sample. Moreover, a nonparametric t-test with 999 random Monte Carlo permutations was used to determine whether there were statistically significant differences in terms of microbial diversity between the infected and non infected mice. β-Diversity among samples was calculated using the UniFrac metric44 and PCoAs were conducted by evaluating both weighted and unweighted Unifrac distances, to highlight eventual clustering of the samples depending on the infection. Data separation in the PCoAs was tested using a permutation test with pseudo-F-ratios. For relative abundance analysis, a Mann–Whitney U-test was used and all statistical evaluations were performed in Matlab (Natick, MA).
The statistical tests for all the other experimental data were performed by use of the GraphPad Prism software (GraphPad Software, La Jolla, CA) and Student’s t-test and Mann–Whitney test for simple comparison or one-way analysis of variance test with Tukey’s correction for multiple comparisons.
References
Beura, L.K. et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016).
Didierlaurent, A. et al. The impact of successive infections on the lung microenvironment. Immunology 122, 457–465 (2007).
Deffur, A. et al. Co-infection with Mycobacterium tuberculosis and human immunodeficiency virus: an overview and motivation for systems approaches. Pathog. Dis. 69, 101–113 (2013).
Chatterjee, S. & Nutman, T.B. Helminth-induced immune regulation: implications for immune responses to tuberculosis. PLoS Pathog. 11, e1004582 (2015).
Potian, J.A. et al. Preexisting helminth infection induces inhibition of innate pulmonary anti-tuberculosis defense by engaging the IL-4 receptor pathway. J. Exp. Med. 208, 1863–1874 (2011).
Rafi, W. et al. 'Coinfection-helminthes and tuberculosis'. Curr. Opin. HIV AIDS 7, 239–244 (2012).
Stewart, G.R. et al. Onchocerciasis modulates the immune response to mycobacterial antigens. Clin. Exp. Immunol. 117, 517–523 (1999).
Hawkes, M. et al. Malaria exacerbates experimental mycobacterial infection in vitro and in vivo. Microbes Infect. 12, 864–874 (2010).
Mueller, A.K. et al. Natural transmission of Plasmodium berghei exacerbates chronic tuberculosis in an experimental co-infection model. PLoS One 7, e48110 (2012).
Cooper, A.M. et al. Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 4, 252–260 (2011).
Antonelli, L.R. et al. Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest. 120, 1674–1682 (2010).
Segura-Lopez, F.K. et al. Association between Helicobacter spp. infections and hepatobiliary malignancies: a review. World J. Gastroenterol. 21, 1414–1423 (2015).
Fox, J.G. et al. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 32, 1238–1245 (1994).
Fox, J.G. et al. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 4, 22–30 (2011).
Taylor, N.S. et al. Enterohepatic Helicobacter species are prevalent in mice from commercial and academic institutions in Asia, Europe, and North America. J. Clin. Microbiol. 45, 2166–2172 (2007).
Boutin, S.R. et al. Different Helicobacter hepaticus strains with variable genomic content induce various degrees of hepatitis. Infect. Immun. 73, 8449–8452 (2005).
Kuehl, C.J. et al. Colonization of the cecal mucosa by Helicobacter hepaticus impacts the diversity of the indigenous microbiota. Infect. Immun. 73, 6952–6961 (2005).
Simpson, R.J. et al. Exercise and the aging immune system. Ageing Res. Rev. 11, 404–420 (2012).
Appelberg, R. et al. Susceptibility of beige mice to Mycobacterium avium: role of neutrophils. Infect. Immun. 63, 3381–3387 (1995).
Beisiegel, M. et al. Combination of host susceptibility and virulence of Mycobacterium tuberculosis determines dual role of nitric oxide in the protection and control of inflammation. J. Infect. Dis. 199, 1222–1232 (2009).
Fulton, S.A. et al. Neutrophil-mediated mycobacteriocidal immunity in the lung during Mycobacterium bovis BCG infection in C57BL/6 mice. Infect. Immun. 70, 5322–5327 (2002).
Keller, C. et al. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect. Immun. 74, 4295–4309 (2006).
Kondratieva, T.K. et al. B cells delay neutrophil migration toward the site of stimulus: tardiness critical for effective bacillus Calmette–Guerin vaccination against tuberculosis infection in mice. J. Immunol. 184, 1227–1234 (2010).
Blomgran, R. & Ernst, J.D Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 186, 7110–7119 (2011).
Zhang, X. et al. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity 31, 761–771 (2009).
Yang, I. et al. Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PLoS One 8, e70783 (2013).
Wu, J. et al. Sputum microbiota associated with new, recurrent and treatment failure tuberculosis. PLoS One 8, e83445 (2013).
Winglee, K. et al. Aerosol Mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PLoS One 9, e97048 (2014).
Perry, S. et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS One 5, e8804 (2010).
Roussos, A. et al. Respiratory diseases and Helicobacter pylori infection: is there a link? Respiration 73, 708–714 (2006).
Torres, M.A. et al. No association between Helicobacter pylori and Mycobacterium tuberculosis infections among gastrointestinal clinic attendees in Lima, Peru. Epidemiol. Infect. 130, 87–91 (2003).
Adriani, A. et al. Helicobacter pylori infection and respiratory diseases: actual data and directions for future studies. Miner. Med. 105, 1–8 (2014).
Arnold, I.C. et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J. Clin. Invest. 121, 3088–3093 (2011).
Suerbaum, S. et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl Acad Sci. USA 100, 7901–7906 (2003).
Lee, A. et al. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112, 1386–1397 (1997).
Sayes, F. et al. Strong immunogenicity and cross-reactivity of Mycobacterium tuberculosis ESX-5 type VII secretion: encoded PE-PPE proteins predicts vaccine potential. Cell Host Microbe 11, 352–363 (2012).
Fox, J.G. et al. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 114, 755–763 (1998).
Ahmadian, A. et al. Pyrosequencing: history, biochemistry and future. Clin. Chim. Acta 363, 83–94 (2006).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acid Res 41, e1 (2013).
Masella, A.P. et al. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinform. 13, 31 (2012).
Caporaso, J.G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R.C Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Wang, Q. et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Lozupone, C. & Knight, R UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Acknowledgements
This work was supported by grants from Institut Pasteur and by the European Union’s Seventh Framework Program (280873 ADITEC; to CL). FS was supported by the University of Damas, Syria and the Programme Transversal de Recherche (No. 441) from Institut Pasteur. RB acknowledges support from the Fondation pour la Recherche Medicale (DEQ20130326471). We gratefully thank Dr G Milon (Institut Pasteur, Paris, France) and Dr L Lo-Man (Institut Pasteur) for helpful advice and discussion, Dr C Josenhans (Hannover Medical School, Hannover, Germany) for sending the Helicobacter hepaticus 3B1 strain, Dr J Jaubert and Dr X Montagutelli (Institut Pasteur) for helpful advices and providing the SEG Mus musculus wild-type mice, E Vimont (Institut Pasteur) for performing Helicobacter-specific PCR and pyrosequencing, E Morillon (Institut Pasteur) for technical assistance, and K Sébastien, E Maranghi, M Szatanik, and I Lanctin (Institut Pasteur) for their excellent technical assistance with animal care or gavages with Helicobacter in the BSL3 animal facilities at the Institut Pasteur. The authors acknowledge the Cytometry Core Facility (Sophie Novault and Pierre-Henri Commere) for their support with flow cytometry.
Author contributions
L.M. designed and performed experiments, analyzed results, and wrote the paper, F.S. performed cytometry and aerosol M. tuberculosis challenges and analyzed results, J.F.B. and E.T. designed and performed experiments involving acquired or induced Helicobacter colonization and analyzed results, A.P. and V.M. performed oral gavages or aerosol challenges, G.J. and M.H. performed histological experiments and analyses, M.S., C.C. and C.P. designed and performed the microbiota experiments, analyzed the results, and wrote the concerned sections, R.B. designed experiments and wrote the paper, and C.L. designed experiments, analyzed results, and wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Majlessi, L., Sayes, F., Bureau, JF. et al. Colonization with Helicobacter is concomitant with modified gut microbiota and drastic failure of the immune control of Mycobacterium tuberculosis. Mucosal Immunol 10, 1178–1189 (2017). https://doi.org/10.1038/mi.2016.140
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2016.140
This article is cited by
-
Gut microbiota modulates bleomycin-induced acute lung injury response in mice
Respiratory Research (2022)
-
Increased Th17 activation and gut microbiota diversity are associated with pembrolizumab-triggered tuberculosis
Cancer Immunology, Immunotherapy (2020)
-
Immunological roulette: Luck or something more? Considering the connections between host and environment in TB
Cellular & Molecular Immunology (2018)
