
Abstract
Hereditary forms of hearing impairment (HI) caused by GJB2 (Cx26) mutations are the frequent sensory disorders registered among newborns in various human populations. In this study, we present data on the molecular, audiological and population features of autosomal recessive deafness 1A (DFNB1A) associated with the donor splicing site IVS1+1G>A mutation of GJB2 gene in Yakut population isolate of the Sakha Republic (Yakutia) located in Eastern Siberia (Russian Federation). The Yakut population exhibits high frequency of some Mendelian disorders, which are rare in other populations worldwide. Mutational analysis of GJB2 gene in 86 unrelated Yakut patients with congenital HI without other clinical features has been performed. In this study, we registered a large cohort of Yakut patients homozygous for the IVS1+1G>A mutation (70 unrelated deaf subjects in total). Detailed audiological analysis of 40 deaf subjects with genotype IVS1+1G>A/IVS1+1G>A revealed significant association of this genotype with mostly symmetrical bilateral severe to profound HI (85% severe-to-profound HI versus 15% mild-to-moderate HI, P<0.05). The highest among six investigated Eastern Siberian populations carrier frequency of the IVS1+1G>A mutation (11.7%) has been found in Yakut population. Reconstruction of 140 haplotypes with IVS1+1G>A mutation demonstrates the common origin of all mutant chromosomes found in Yakuts. The age of mutation was estimated to be approximately 800 years. These findings characterize Eastern Siberia as the region with the most extensive accumulation of the IVS1+1G>A mutation in the world as a result of founder effect.
Similar content being viewed by others


Introduction
Congenital deafness is one of the most frequent sensory disorders which accounts for about 1 in 1000 newborns, and approximately half of all cases have a genetic etiology.1 Hereditary forms of hearing impairment (HI) are characterized by clinical polymorphism and genetic heterogeneity. To date, 114 loci and 55 genes have been described for inherited non-syndromic HI. About 75% of non-syndromic deafness cases are inherited as an autosomal recessive trait, 10 to 15%—as autosomal dominant, low portion are X-linked and some cases are caused by mitochondrial DNA mutations.2 One of the main forms of non-syndromic HI is autosomal recessive deafness 1A (DFNB1A, MIM ID#220290) caused by mutations in gene GJB2 (13q11–q12), including digenic (GJB2/GJB6 and GJB2/GJB3) mutations in two genes GJB6 and GJB3 in chromosomal regions 13q12 and 1p35.1, respectively.3 The genes GJB2, GJB6 and GJB3 encode gap-junction proteins connexins 26, 30 and 31, respectively, that oligomerize to hexamers to form transmembrane channels for the potassium ions being recycled across supporting cells in an inner ear. Main cause of DFNB1A in various human populations are GJB2 mutations, and GJB6 and GJB3 mutations are less prevalent.4, 5 To date, about 150 different mutations (mostly recessive), polymorphic variants and changes in nucleotide sequence of GJB2 gene with unknown relation to the disease have been reported.6 The mutation c.35delG has been found to occur with high frequency in populations of Europe, Middle East and Northern America (predominantly among Caucasians).7, 8, 9, 10, 11, 12, 13, 14 The mutation c.235delC has been registered mainly in East Asian populations (Japanese, Korean and Chinese); and also has been found among Thais (Southeast Asia), Mongolians (Central Asia) and Altaians (South Siberia).15, 16, 17, 18, 19, 20, 21 The mutation c.167delT is specific for Ashkenazi Jews and is sporadically found in some other populations.22 Mutation p.Trp24X is widespread in India23 and in Romany (Gypsy) population (Eastern Europe).24 Mutation p.Arg143Trp is a major mutation in Ghana (West Africa),25 and mutation p.Val37Ile has the greatest prevalence among the populations of Southeast Asia (Thailand).21
In all, 15–20 cases of congenital/early onset HI in 14 000–15 000 live births (∼1:900–1:750 newborns) are detected per year in the Sakha Republic (Yakutia) located in Eastern Siberia (that is an extensive Asian part of territory of the Russian Federation). According to the epidemiological data, high rate of congenital HI is caused by some hereditary forms of deafness spread in indigenous populations of Sakha Republic.26 The main indigenous people of the Sakha Republic and the largest aboriginal population of Siberia is represented by Turkic-speaking Yakuts (originally named as the Sakha), whose population amounts to 432 290. The Yakuts are characterized by specific anthropological, demographic, linguistic and historical features indicative of their relationships to nomadic Turkic tribes of South Siberia and Central Asia.27, 28 The genetic data revealed a relatively small size of Yakut ancestor population and the strong bottleneck effect in the Yakut paternal line (∼80% of Y chromosomes of Yakuts belong to one haplogroup—N1c).29, 30 Marriage traditions and geographical isolation had a significant role in genetic and demographic history of the Yakut population.31 Moreover, high frequency of some Mendelian disorders in the Yakut population was found to be a result of founder effect.32, 33, 34, 35, 36 Preliminary mutational analysis of the coding region of GJB2 gene in deaf patients from the Sakha Republic revealed the presence of GJB2 mutations in 50.1% of Caucasian patients (Russians, Ukrainians, Ingush) and only in 7.2% of Asian patients (predominantly Yakuts).37 We suggested that high prevalence of congenital HI in the Sakha Republic may be caused by mutations in non-coding region of GJB2 gene or in other genes responsible for hereditary forms of deafness among Yakuts.
In this study, we present data on the molecular, audiological and population features of autosomal recessive deafness 1A (DFNB1A) associated with the donor splicing site IVS1+1G>A mutation of GJB2 gene in the Sakha Republic.
Materials and methods
Patients
Data on individuals with HI were obtained from the Republican Hospital No 1, National Medical Centre, Ministry of Public Health of the Sakha Republic, and collected from the students of the Republican special residential schools for the deaf and hearing impaired children (Yakutsk, Russian Federation). We ascertained 114 patients with HI. In all, 28 subjects were excluded from the study, as they had syndromic, unclassified forms of HI, as HI associated with malformations, or HI due to evident environmental causes (Supplementary table). Among 86 unrelated patients with non-syndromic hearing loss included in the study, 46 (53.4%) individuals were males and 40 (46.5%) were females. All patients were Yakuts aged 0–44, in average 17±9.05 years old. Autosomal recessive inheritance of the disease occurred in 30 families, including 11 families with affected siblings and 19 with other relatives. Inheritance type of HI was not identified in 47 families due to the absence of known cases of deafness among the relatives, hence these cases were considered as sporadic. Assortative marriages between deaf parents were observed in nine families, and in these cases, the type of inheritance could not be identified. Therefore, the clinical and genealogical analysis demonstrated mainly autosomal recessive inheritance of disease and the absence of obvious autosomal dominant forms of HI among examined patient's group.
Audiological analysis
We collected audiometric data on 40 patients with genotype IVS1+1G>A/IVS1+1G>A. Audiograms from available medical documents were analyzed retrospectively for each ear separately. All 40 patients (80 ears) underwent otoscopic examination, tympanometry (Tympanometer AT-235, Interacoustics, Assens, Denmark) and audiometric testing. In most cases (34 patients), hearing thresholds were determined by pure-tone audiometry, using a clinical tonal audiometer GSI 60 (Grason-Stadler, Madison, WI, USA) in a soundproof room according to current clinical standards. Hearing thresholds for six patients (children aged 0–6 years) were determined by electrophysiological measurements of the auditory steady-state response and auditory evoked potential using ‘GSI Audera’ (Grason-Stadler). Air-conduction thresholds were obtained at 0.125, 0.25, 0.5, 1, 2, 4 and 8 kHz. Severity of hearing loss was defined as mild (25–40 dB), moderate (41–70 dB), severe (71–90 dB) or profound (above 90 dB). Data on hearing thresholds for 49 ears of patients with hearing ability up to 4–8 kHz were used for detailed analysis.
Mutation analysis of GJB2 gene in patients
The blood samples were obtained from 86 unrelated Yakut patients with non-syndromic HI, and from their siblings and/or their parents. The genomic DNA was extracted from lymphocytes of peripheral blood. Amplification of the coding exon 2 and flanking intronic regions was performed using the following primers: Cx26A-U/Cx26U-L (forward 5′-TCTTTTCCAGAGCAAACCGC-3′, reverse 5′-GACACGAAGATCAGCTGCAG-3′) (285 bp),7 Cx342U/Cx739-L (forward 5′-AGGCCGACTTTGTCTGCAACA-3′, reverse 5′-GTGGGCCGGGACACAAAG-3′) (415 bp),9 5′-TATGTCATGTACGACGGCT-3′, 5′-TCTAACAACTGGGCAATGC-3′ (239 bp).8 Amplification of the non-coding exon 1 and flanking intronic regions was performed using primers Ex1-F/Ex1-R (forward 5′-CCGGGAAGCTCTGAGGAC-3′, reverse 5′-GCAACCGCTCTGGGTCTC-3′), with the addition of 10% Betaine (Sigma-Aldrich, St Louis, MO, USA).38 The products of PCR were subjected to direct sequencing using the same primers on ABI PRISM 3130XL (Applied Biosystems, USA).
Carrier frequency of IVS1+1G>A mutation
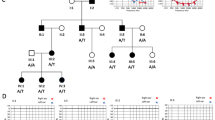
Molecular screening for mutation IVS1+1G>A of GJB2 gene was performed using PCR–restriction fragment length polymorphism analysis (HphI). In all, 423 DNA samples of hearing individuals from six populations of Yakutia, Yakuts (n=120), Dolgans (n=43), Evenks (n=80), Evens (n=50), Yukaghirs (n=50) and Russians (n=80), were obtained from the DNA Bank of the Department of Molecular Genetics of Yakut Research Center of Complex Medical Problems of RAMS (Yakutsk, Russian Federation). These samples were tested only for either presence or absence of the IVS1+1G>A mutation as described in legend to Figure 1. The results of this testing remained completely anonymous.

Detection of the IVS1+1G>A mutation. (a) Sequencing results: normal person, wild type (Wt)/Wt (upper panel); heterozygote for the IVS1+1G>A (middle panel), and homozygote for the IVS1+1G>A (lower panel); (b) detection of GJB2 mutation IVS1+1G>A in 4% agarose gel by PCR–restriction fragment length polymorphism analysis with use of HphI. The mutation IVS1+1G>A abolishes the restriction site for HphI. The homozygous patients (P1 and P2) show undigested 363 bp fragment; two normal hearing siblings (S1 and S2) present two fragments, 293 bp and 70 bp (70 bp fragment is invisible in this gel); heterozygous mother and father of patients show three fragments, 363, 293 bp, and 70 bp (70 bp fragment is invisible in this gel); Ma, marker puc19/MspI; (c) pedigree of family with IVS1+1G>A and sequencing data.
Haplotype analysis
DNA samples of 70 unrelated deaf patients homozygous for IVS1+1G>A mutation and DNA samples of 106 unrelated individuals without this mutation were used for the haplotype analysis. All patients were Yakuts originated from various regions of Sakha Republic. Eight polymorphic microsatellite markers: D4S189, D13S1316, D13S141, D13S175, D13S1853, D13S143, D13S1275, D13S292 and two single-nucleotide polymorphism (SNPs), rs2274083 (p.Val27Ile) and rs2274084 (p.Glu114Gly), were used for the linkage disequilibrium analysis. All markers and primers were selected using the appropriate databases Enseble dated June–July 2009 (http://www.ensembl.org/index.html) and NCBI dated June–July 2009 (http://www.ncbi.nlm.nih.gov/unists/). The amplified products were resolved on 10% polyacrylamide gel with ethidium bromide staining under ultraviolet light to verify size and quantity. Genotyping of SNPs, rs2274083 (p.Val27Ile) and rs2274084 (p.Glu114Gly), was performed using PCR–restriction fragment length polymorphism analysis (Fok I and Tas I).21 The PCR–restriction fragment length polymorphism products were resolved on 8% polyacrylamide gel with ethidium bromide staining under ultraviolet light to verify size and quantity. The total physical distance of the GJB2 gene region flanking with examined markers was ∼4.6 Mb base pair (Figure 4).
Mutation age
The age of the mutation IVS1+1G>A was estimated using the following equation

where Q is an observed frequency of disease chromosomes not carrying the progenitor marker allele; Pn, an observed frequency of marker allele in normal chromosomes; q, a number of generations; and θ, a recombination rate. For estimation of mutation age, we have used data on the distal marker D13S143 linkage with founder haplotype. On the basis of distance from D13S143 to IVS1+1G>A (∼1.5 Mb), and considering a linear relationship of 1 cM=1000 kb, the calculated recombination rate (θ) is 0.015 per generation. Values of the parameters used were Q=0.062, Pn=0.84, θ=0.015. Generation time was estimated to be 25 years.
Statistical analyses
Differences in IVS1+1G>A mutation frequencies between the studied groups (95% credible interval) were computed with the program ‘Sampling’ kindly provided by V Macaulay and adapted by M Metspalu (Estonian Biocentre, Tartu, Estonia). Differences between groups of examined eight polymorphic microsatellite and two SNPs markers were tested with χ2-statistics. P-values are <0.05 were considered as statistically significant. To evaluate the linkage disequilibrium, we used the algorithm suggested earlier.39
Results
Identification of donor splicing site GJB2 mutation IVS1+1G>A in Yakut patients
The non-coding exon 1 and coding exon 2 and flanking regions of GJB2 gene were analyzed by sequencing. No mutations of GJB2 gene were found in 10 patients. In 70 out of 86 unrelated patients with non-syndromic HI, we found a homozygous G>A transition in intron 1 known as donor splicing site −3170 G>A (IVS1+1G>A) mutation corresponding to the AUG translation-initiating codon.40, 41 Six patients were compound heterozygotes for IVS1+1G>A and other mutations or polymorphisms in coding region of GJB2 gene (c.35delG, p.Val27Ile, p.Val27Ile+p.Glu114Gly). Detection of the IVS1+1G>A mutation is shown in Figure 1.
Audiological analysis of 40 patients with genotype IVS1+1G>A/IVS1+1G>A
Audiometric examinations of 40 patients homozygous for the IVS1+1G>A mutation were based on the retrospective analysis from the available medical documents. In all, 85% of patients demonstrate severe to profound HI; 14%, moderate and 1%, mild hearing loss (Figure 2a). A slight asymmetry between two ears of each patient (difference in pure-tone averages (PTA)0.5, 1, 2, 4 kHz⩽15 dB) was found in eight patients (20%), and difference in PTA0.5, 1, 2, 4 kHz between two ears equal to 20 dB was observed only in three patients (7.5%). According to their audiological characteristics, we subdivided all examined patients into two subgroups. First subgroup included 49 out of 80 examined ears, which demonstrated the hearing ability in the range from 0.125 to 4 kHz (all 49 ears) or from 0.125 to 8 kHz (30 ears) with high variability of hearing thresholds on each test frequency (Figure 2b). To demonstrate the hearing thresholds of this patient subgroup in more details, we have constructed scatter diagrams showing the binaural mean PTA0.5, 1, 2, 4 kHz for data on 49 ears of patients (median 86 dB; Figure 3). Hearing thresholds with the range of 90–115 dB were detected in approximately one-half of examined patients (23 ears out of 49 ears), with the range of 70–90 dB in 14 ears, with the range of 40–70 dB in 11 ears and 25–40 dB in one ear. Second subgroup included 31 out of 80 ears with hearing ability in the range from 0.125 to 2 kHz. All 31 examined ears demonstrated the hearing ability in the range from 0.125 to 0.5 kHz; 28 ears, from 0.125 to 1 kHz; and only 13 ears, from 0.125 to 2 kHz. Hearing threshold averages in this subgroup were no <75 dB on each test frequency. Moreover, variability of hearing thresholds in this subgroup was considerably less than in first subgroup (Figure 2c).

Audiometric data on 40 patients (80 ears) with genotype IVS1+1G>A/IVS1+1G>A. (a) Relative frequencies of HI levels in examined patients were mild, 25–40 dB; moderate; 41–70 dB; severe, 71–90 dB; and profound, above 90 dB. (b) Audiogram of average values for first subgroup (49 ears): hearing ability was registered in wide range of frequencies (from 0.125 to 4–8 kHz); (c) audiogram of average values for second group (31 ears): hearing ability was registered from 0.125 up to 2 kHz, arrow shows frequency limits of hearing (up to 0.5 kHz, 31 ears; 1 kHz, 28 ears; 2 kHz, 13 ears). Bold points denote average values of hearing thresholds, transparent points denote maximum and minimum values of hearing thresholds.

Scatter diagram of the PTA0.5, 1, 2, 4 kHz in first subgroup (49 ears) of patients with hearing ability ranged up to 4–8 kHz. Each ear was shown as open circle. Dotted lines demarcate hearing thresholds: mild HI (from 0 to 40 dB), moderate HI (from 41 to 70 dB), severe HI (from 71 to 90 dB) and profound HI (from 91 to 120 dB). Median of hearing thresholds is 86 dB (severe HI).
Carrier frequency of IVS1+1G>A in six populations of Eastern Siberia
Extremely high prevalence of splice site mutation IVS1+1G>A, observed in homozygous state in Yakut deaf patients allowed us to propose that IVS1+1G>A may also be a common pathogenic mutation of GJB2 gene among other Northeast Asians. However, Siberian populations are significantly distinguished by anthropologic and linguistic affiliations, as well as by their population genetic history. We studied carrier frequency of IVS1+1G>A mutation in several indigenous populations of Sakha Republic (Eastern Siberia): Turkic-speaking Yakuts and Dolgans, Tungusic-speaking Evenks and Evens, and Yukaghirs with uncertain (Paleo-Asiatic or Uralic) linguistic affiliation, as well as Slavic-speaking Russians inhabiting the Sakha Republic (Table 1). Among 423 individuals with normal hearing, originated from six investigated populations, mutation IVS1+1G>A in heterozygous state was found in 20 subjects: Yakuts (from Central and Vilyuy subpopulations; 14/120), Dolgans (2/43), Evenks (3/80) and Evens (1/50). This mutation was not found in Yukaghirs (0/50) and Russians (0/80; Table 1). The extremely high carrier frequency of IVS1+1G>A mutation was found in Yakut population (11.7%).
Founder haplotype and age of mutation IVS1+1G>A
To test the hypothesis whether mutation IVS1+1G>A in Yakuts has a common origin, we have performed a haplotype analysis by studying eight polymorphic microsatellite and two SNP markers on 140 chromosomes with IVS1+1G>A mutation and on 212 chromosomes of normal hearing individuals without this mutation (Figure 4). We found linkage disequilibrium for seven microsatellite markers D4S189, D13S1316, D13S141, D13S175, D13S1853, D13S143, D13S1275, and one SNP rs2274083 (p.Val27Ile) marker. The distal markers, rs2274083 (p.Val27Ile), D13S1316, D13S141, D13S175, D13S1853, and D13S143, showed strong association (P<0.05) versus the proximal markers, D4S189, D13S1275 and D13S292, at GJB2 gene (Figure 4). According to χ2-values and linkage disequilibrium parameter, we proposed that a founder haplotype consists of alleles D4S189(3)-D13S1316(2)-D13S141(5)-p.Val27Ile(G)-D13S175(3)-D13S1853(4)-D13S143(1) (Figure 4). This haplotype was ascertained for eight chromosomes with the mutation IVS1+1G>A and was not found among chromosomes without this mutation. The structure of identified haplotypes indicates the common origin of all studied mutant chromosomes. The results are shown in Figure 4. Divergence time of the founder haplotype with the mutation IVS1+1G>A is estimated to be about 800 years. The period of expansion of chromosomes with IVS1+1G>A mutation in Yakutia has been dated approximately to the 13th century AD.

Haplotype analysis. (a) Physical map of microsatellite and SNP markers flanking GJB2 gene. (b) Haplotypes of 140 chromosomes with splice site IVS1+1G>A mutation of GJB2 gene of 70 unrelated Yakut patients with DFNB1A. Two alleles of examined markers are separated by slash. Putative founder haplotype is shaded in gray. PB, patient's places of birth; C, Central districts; V, Vilyuy districts; N, Northern districts; S, Southern districts of the Republic of Sakha; Y, Yakutsk city. The places of birth of patients with recombinant founder haplotype are highlighted in bold and denoted by asterisks (C*, V*, Y*). Yak, Yakut.
Discussion
The first study of molecular, audiological and population features of autosomal recessive deafness (DFNB1A) associated with the donor splicing site mutation IVS1+1G>A in GJB2 gene was performed in Yakut population of the Sakha Republic (Eastern Siberia). We revealed 70 homozygotes and 6 compound heterozygotes for the mutation IVS1+1G>A. Interestingly, a c.235delC mutation, the most common pathogenic allelic variant in East Asian population,15, 16, 17, 18, 19, 20 was not registered in Yakuts. The mutation IVS1+1G>A accounts for up to about 95% of all deleterious mutations in GJB2 gene, which have been found in examined Yakut patients. This finding characterizes Eastern Siberia as the region with the most extensive accumulation of this mutation in the world. According to previous results, the sequences of complementary DNA from lymphoblastoid cell line from patients with genotype IVS1+1G>A/c.35delG yielded a message only from the c.35delG allele, indicating that the IVS1+1G>A allele was not transcribed.14 This finding suggests the pathogenic role of this splice site mutation. However, the audiological features of patients with IVS1+1G>A mutation in homozygous state were not previously investigated in large cohort of patients. Analysis of the audiological characteristics obtained from 40 subjects with genotype IVS1+1G>A/IVS1+1G>A revealed significant association of this genotype with severe to profound HI (85% of severe-to-profound versus 15% of mild-to-moderate, P<0.05) with mostly symmetrical bilateral hearing loss (29 out of 40 examined patients, P<0.05). Moreover, detailed audiological analysis showed a variability in hearing thresholds on different frequency ranges among subjects homozygous for IVS1+1G>A mutation (Figures 2b and c).
Taking into account that a large cohort of individuals homozygous for IVS1+1G>A mutation has been found among the Siberian indigenous people living in the region with extremely severe climate, we do not except that observed phenotypic variability may reflect the effect of environmental factors and/or modifier genes leading to incomplete penetrance and variable expression of genotypes IVS1+1G>A/IVS1+1G>A.
Autosomal recessive deafness 1A (DFNB1A) is not the first inherited disease registered with high rate among the Yakut population known as the population isolate in Asia.33 The Yakut population exhibits high rate of some Mendelian disorders, which are rare in other populations worldwide. High rates of spinocerebellar ataxia type 1 (SCA1),32 myothonic dystrophy (DMPK),26 oculopharyngeal muscular dystrophy (OPMD),34 autosomal recessive methemoglobinemia (DIA1)35 and two types of short stature disorders, 3-M (CUL7) and short stature with optic atrophy and Pelger-Huët anomaly (SOPH) syndrome (NBAS),33, 36 were previously found in Yakut population. We estimated the prevalence of DFNB1A associated with splice site IVS1+1G>A mutation in GJB2 gene as 16.2 in 100 000 in Yakut population, that is, higher compared with other common autosomal reccesive disorders found in Yakuts, such as autosomal recessive methemoglobinemia (14.9 in 100 000),35 two types of short stature disorders, 3-M (10.0 in 100 000),33 and SOPH syndromes (9.95 in 100 000).36 Thus, we suggested that autosomal recessive deafness 1A (DFNB1A) associated with homozygous mutation IVS1+1G>A in GJB2 gene is the most common autosomal recessive disease among Yakuts.
Carrier frequency of IVS1+1G>A is apparently associated with specific linguistic affiliation of studied ethnical groups (Table 1). High carrier frequency of this mutation was revealed in Turkic-speaking populations of Yakuts (11.7%) and Dolgans (4.7%). Lower rate of this mutation was found in Tungusic-speaking populations of Evenks (3.8%) and Evens (2.0%), and this mutation was not found at all in Uralic or Paleo-Asiatic-speaking Yukaghirs and Slavic-speaking Russians.
The highest among other studied Eastern Siberian populations carrier frequency of the IVS1+1G>A mutation (11.7%) was found in isolated Yakut population. Similar data on some autosomal recessive disorders were obtained for other human population isolates. It was found that carrier frequency of the E143 K (c.1505G>A) mutation in CYP1B1 gene associated with primary congenital glaucoma equals 10.8% in Slovak Gypsies (Roms).42 In Ashkenazi Jewish carrier frequencies of recessive mutations were as follows: Gaucher disease, 1/15 (6.7%); cystic fibrosis, 1/24 (4.2%); and Tay-Sachs disease, 1/28 (3.6%).43 In a small Muslim Arab village, 15% of healthy individuals were carriers of three GJB2 mutations.44
On the basis of observed frequency of heterozygotes (11.7% of 432 290 people) for the IVS1+1G>A mutation in the Yakut population, we calculated an expected number of homozygotes, which was approximately 1400 affected subjects (0.3% of whole population). This calculation indicates that the prevalence of DFNB1A due to the IVS1+1G>A mutation in the Sakha Republic may be even higher than observed estimation of 16.2 in 100 000.
Discrepancy between expected and observed rates of DFNB1A deafness due to the IVS1+1G>A in the Yakut population could be a consequence of some peculiarities of the Yakut population structure and demographic factors. Density of the Yakut population was always low, and the majority of the Yakut population was dispersed in widely separated, relatively small settlements. Subdivided Yakut population is characterized by different rates of mutual migrations depending on remoteness of local settlements. Rates of endogamy and interethnic matings correlate with the size and ethnic composition of local subpopulations of Yakuts. Consanguineous marriages were traditionally uncommon in the Yakut population, and the total inbreeding estimated by isonymy (F(it)) varies from 0 to 0.007576.45 All these features taken together could influence the pattern of the DFNB1A prevalence associated with IVS1+1G>A mutation in the Yakut population.
The founder effect is the most plausible explanation for the increased incidence of some rare diseases and high carrier frequency of single pathogenic mutations in most of human population isolates. Reconstruction of 140 haplotypes with IVS1+1G>A mutation demonstrates the common origin of all mutant chromosomes found in Yakuts. Highest diversity of haplotypes was found in the Central and Vilyuy sub-populations of Yakuts (excluding of the Yakutsk city), indicating that the expansion of mutant chromosomes on the territory of the Sakha Republic had started from the Lena-Amga interfluves area (Central district) and the Vilyuy river basin (Vilyuy district) (Figure 5). These data correspond to known historical facts about initial settling of Yakuts in Central and Vilyuy regions, and their later expansion to the Northern part of Yakutia.46 The calculated age of mutation (∼800 years) correlates with last migration of Turkic-speaking Yakut ancestors in East Siberia in 13th–14th centuries AD.28 Previous studies demonstrated that mutation IVS1+1G>A was predominantly registered among Caucasian populations.14, 38, 40, 41, 47, 48, 49 Relatively high rates of this mutation in Eastern Europe (Czech Republic, Poland and Hungary) have been supposed to be specific for Slavic populations.47 In recent study of Tekin et al.,50 identification of this mutation in homozygous state in Turks and Mongolians and reconstruction of common haplotype with IVS1+1G>A proposes Central Asian origin for this mutation followed by the migration of its carriers to the territories of Middle East.50 The data on high prevalence of IVS1+1G>A in Northeastern Asians, particularly in Yakuts, may provide an additional evidence for common Central Asian origin of this mutation, because it was supposed that Turkic-speaking ancestors of Yakuts migrated to the Eastern part of Siberia from their initial settlement in the area of the Baikal Lake under pressure of the Mongol expansion beginning from the 13th century AD.28, 29 Currently, data on the prevalence of IVS1+1G>A is very scarce, particularly for Asian populations. Therefore, for estimation of the geographical area of the IVS1+1G>A mutation prevalence, its possible common founder effect, and migration routes of the IVS1+1G>A mutation carriers in the world, further extensive studies in many populations are required.

The geographic distribution of the founder haplotype with mutation IVS1+1G>A in the Republic of Sakha. Ancient founder haplotype with the IVS1+1G>A and recombinant haplotypes with this mutation are denoted as small open and closed circles, correspondingly. Central districts of the Republic of Sakha are shown in circle on the right and Vilyuy districts in circle on the left. Arrows show the possible migration routes of founder haplotype with IVS1+1G>A.
References
Marazita, M. L., Ploughman, L. M., Rawlings, B., Remington, E., Arnos, K. S. & Nance, W. E. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med. Genet. 46, 486–491 (1993).
Van Camp, G. & Smith, R. J. H. Hereditary Hearing Loss Homepage, July–August 2010; http://hereditaryhearingloss.org.
Mignon, C., Fromaget, C., Mattei, M. G., Gros, D., Yamasaki, H. & Mesnil, M. Assignment of connexin 26 (GJB2) and 46 (GJA3) genes to human chromosome 13q11-q12 and mouse chromosome 14D1-E1 by in situ hybridization. Cytogenet. Cell. Genet. 72, 185–186 (1996).
del Castillo, F. J., Rodríguez-Ballesteros, M., Álvarez, A., Hutchin, T., Leonardi, E., de Oliveira, C. A. et al. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 42, 588–594 (2005).
Liu, X. Z., Yuan, Y., Yan, D., Ding, E. H., Ouyang, X. M., Fei, Y. et al. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum. Genet. 125, 53–62 (2009).
Ballana, E., Ventayol, M., Rabionet, R., Gasparini, P. & Estivill, X. Connexins and Deafness Homepage, July–August 2010; http://davinci.crg.es/deafness/.
Kelsell, D. P., Dunlop, J., Stevens, H. P., Lench, N. J., Liang, J. N., Parry, G. et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387, 80–83 (1997).
Zelante, L., Gasparini, P., Estivill, X., Melchionda, S., D′Agruma, L., Govea, N. et al. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 6, 1605–1609 (1997).
Kelley, P. M., Harris, D. J., Comer, B. C., Askew, J. W., Fowler, T., Smith, S. D. et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am. J. Hum. Genet. 62, 792–799 (1998).
Gasparini, P., Rabionet, R., Barbujani, G., Melçhionda, S., Petersen, M., Brøndum-Nielsen, K. et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur. J. Hum. Genet. 8, 19–23 (2000).
Rabionet, R., Zelante, L., López-Bigas, N., D′Agruma, L., Melchionda, S., Restagno, G. et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 gene. Hum. Genet. 106, 40–44 (2000).
Lucotte, G., Bathelier, C. & Champenois, T. PCR test for diagnosis of the common GJB2 (connexin 26) 35delG mutation on dried blood spots and determination of the carrier frequency in France. Mol. Cell. Probes 15, 57–59 (2001).
Tekin, M., Akar, N., Cin, S., Blanton, S. H., Xia, X. J., Liu, X. Z. et al. Connexin 26 (GJB2) mutations in the Turkish population: implications for the origin and high frequency of the 35delG mutation in Caucasians. Hum. Genet. 108, 385–389 (2001).
Shahin, H., Walsh, T., Sobe, T., Lynch, E., King, M. C., Avraham, K. B. et al. Genetics of congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum. Genet. 110, 284–289 (2002).
Park, H. J., Hahn, S. H., Chun, Y. M., Park, K. & Kim, H. N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 110, 1535–1538 (2000).
Liu, X. Z., Xia, X. J., Ke, X. M., Ouyang, X. M., Du, L. L., Liu, Y. H. et al. The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum. Genet. 111, 394–397 (2002).
Ohtsuka, A., Yuge, I., Kimura, S., Namba, A., Abe, S., Van Laer, L. et al. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum. Genet. 112, 329–333 (2003).
Posukh, O., Pallares-Ruiz, N., Tadinova, V., Osipova, L., Claustres, M. & Roux, A. F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 6, 12 (2005).
Han, S. H., Park, H. J., Kang, E. J., Ryu, J. S., Lee, A., Yang, Y. H. et al. Carrier frequency of GJB2 (connexin-26) mutations causing inherited deafness in the Korean population. J. Hum. Genet. 53, 1022–1028 (2008).
Dai, P., Yu, F., Han, B., Liu, X., Wang, G., Li, Q. et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 14, 7–26 (2009).
Wattanasirichaigoon, D., Limwongse, C., Jariengprasert, C., Yenchitsomanus, P. T., Tocharoenthanaphol, C., Thongnoppakhun, W. et al. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing–impaired and control Thai individuals. Clin. Genet. 66, 452–460 (2004).
Morell, R. J., Kim, H. J., Hood, L. J., Goforth, L., Friderici, K., Fisher, R. et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N. Engl. J. Med. 339, 1500–1505 (1998).
RamShankar, M., Girigajan, S., Dagan, O., Ravi Shankar, H. M., Jalvi, R., Rangasayee, R. et al. Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J. Med. Genet. 40, e68 (2003).
Minárik, G., Ferák, V., Feráková, E., Ficek, A., Poláková, H. & Kádasi, L. High frequency of GJB2 mutation W24X among Slovak Romany (Gypsy) patients with non-syndromic hearing loss (NSHL). Gen. Physiol. Biophys. 22, 549–556 (2003).
Hamelmann, C., Amedofu, G. K., Albrecht, K., Muntau, B., Gelhaus, A., Brobby, G. W. et al. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 18, 84–85 (2001).
Tarskaia, L. A., Zinchenko, R. A., El’chinova, G. I., Egorova, A. G., Korotov, M. N., Basova, E. V. et al. The structure and diversity of hereditary pathology in Sakha Republic (Yakutia). Russ. J. Genet. 40, 1530–1539 (2004).
Alexeev, V. P. & Gokhman, I. I. Anthropology of the Asian part of the USSR (Izdatel'stvo Nauka, Moscow, 1984) (in Russian).
Gogolev, A. I. Ethnical History People of the Yakutia (to Beginning XX Century) (Izdatel'stvo YNC SO RAN, Yakutsk, 2004) (in Russian).
Pakendorf, B., Novgorodov, I. N., Osakovskij, V. L., Danilova, A. P., Protod′jakonov, A. P. & Stoneking, M. Investigating the effects of prehistoric migrations in Siberia: genetic variation and the origins of Yakuts. Hum. Genet. 120, 334–353 (2006).
Fedorova, S. A. & Khusnutdinova, E. K. Gene pools of peoples from the Republic Sakha (Yakutia): structure, origin, genetic relationships. Russ. J. Genet. 46, 1244–1246 (2010).
Tarskaya, L. A., El’chinova, G. I. & Vinokurov, V. N. Analysis of Marital Migrations in two regions in the Sakha Republic (Iakutia). Russ. J. Genet. 39, 1698–1701 (2003).
Lunkes, A., Goldfarb, L. G., Platonov, F. A., Alexeev, V. P., Duenas-Barajas, E., Gajdusek, D. C. et al. Autosomal dominant spinocerebellar ataxia (SCA) in Siberian founder population: assignment to the SCA1 locus. Exp. Neurol. 126, 310–312 (1994).
Maksimova, N., Hara, K., Miyashia, A., Nikolaeva, I., Shiga, A., Nogovicina, A. et al. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: new population isolate in Asia. J. Med. Genet. 44, 772–778 (2007).
Maksimova, N. R., Nikolaeva, I. A., Korotov, M. N., Ikeuchi, T., Onodera, O., Nishizava, M. et al. The clinical-genealogic and molecular-genetic characteristics of oculopharyngeal muscular dystrophy in the Republic of Sakha (Yakutia). Zh. Nevrol. Psikhiatr. Im S S Korsakova 108, 52–60 (2008) (in Russian).
Galeeva, N. M., Nazarenko, L. P., Nazarenko, S. A., Tverskaya, S. M. & Polyakov, A. V. Molecular-genetic cause of recessive congenital methemoglobinemia type 1 in Yakutia. Med. Genet. 9, 15–21 (2005) (in Russian).
Maksimova, N., Hara, K., Nikolaeva, I., Chun-Feng, T., Usui, T., Takagi, M. et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterized by optic nerve atrophy and Pelger-Huët anomaly. J. Med. Genet. 47, 538–548 (2010).
Barashkov, N. A., Dzhemileva, L. U., Fedorova, S. A., Maksimova, N. R. & Khusnutdinova, E. K. Connexin gene 26 (GJB2) mutations in patients with hereditary non-syndromic senserineural loss of hearing in the Sakha Republic (Yakutia). Vestn. Otorinolaringol. 5, 23–28 (2008) (in Russian).
Sirmaci, A., Akcayoz-Duman, D. & Tekin, M. The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J. Genet. 85, 213–216 (2006).
Bengtsson, B. O. & Thompson, G. Measuring the strength of associations between HLA antigens and diseases. Tissue Antigens 18, 356–363 (1981).
Denoyelle, F., Weil, D., Maw, M., Wilcox, S. A., Lench, N. J., Allen-Powell, D. R. et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum. Mol. Genet. 12, 2173–2177 (1997).
Green, G. E., Scott, D. A., McDonalld, J. M., Woodworth, G. G., Sheffield, V. C. & Smith, R. J. Carrier rates in the Midwestern United States for GJB2 mutations causing inherited deafness. JAMA 281, 2211–2216 (1999).
Plásilová, M., Stoilov, I., Sarfarazi, M., Kádasi, L., Feráková, E. & Ferák, V. Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J. Med. Genet. 36, 290–294 (1999).
Kronn, D., Jansen, V. & Ostrer, H. Carrier screening for cystic fibrosis, Gaucher disease, and Tay-Sachs disease in the Ashkenazi Jewish population: the first 1000 cases at New York University Medical Center, New York, NY. Arch. Intern. Med. 158, 777–781 (1998).
Zlotogora, J., Carasquillo, M., Barges, S., Shalev, S. A., Hujerat, Y. & Chakravarti, A. High incidence of deafness from three frequent connexin 26 mutations in an isolated community. Genet. Test 10, 40–43 (2006).
Kucher, A. N., Danilova, A. L., Koneva, L. A. & Nogovitsina, A. N. Marriage structure of Yakut populations: ethnic composition and isonymy inbreeding. Russ. J. Genet. 46, 362–370 (2010).
Dolgikh, B. O. Relationships and tribes of Siberian people in XVII c (Izdatel'stvo Nauka, Moskow, 1960) (in Russian).
Seeman, P. & Sakmaryová, I. High prevalence of IVS1+1 to G>A/GJB2 mutation among Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2. Clin. Genet. 69, 410–413 (2006).
Tóth, T., Kupka, S., Haack, B., Fazakas, F., Muszbek, L., Blin, N. et al. Coincidence of mutations in different connexin genes in Hungarian patients. Int. J. Mol. Med. 20, 315–321 (2007).
Pollak, A., Skórka, A., Mueller-Malesiñska, M., Kostrzewa, G., Kisiel, B., Waligóra, J. et al. M34T and V37I mutations in GJB2 associated hearing impairment: evidence for pathogenicity and reduced penetrance. Am. J. Med. Genet. A 143A, 2534–2543 (2007).
Tekin, M., Xia, X. J., Erdenetungalag, R., Cengiz, F. B., White, T. W., Radnaabazar, J. et al. GJB2 mutations in Mongolia: complex alleles, low frequency, and reduced fitness of the deaf. Ann. Hum. Genet. 74, 155–164 (2010).
Acknowledgements
We thank all patients and blood sample donors who have contributed to this study. Special thanks to physicians from the Medical-Genetic Consulting and the Audiology-Logopaedic Center, the Republican Hospital #1, the Ministry of Public Health of the Sakha Republic (Yakutsk): Nogovicina AN, Maksimova NR, Sukhomyasova AL, Kononova SK, Alexeeva SP, Gurinova EE, Nikolaeva IA, Sivtseva LN and Egorova VN; to specialists of the MK Ammosov North-Eastern Federal University (Yakutsk): Dr Prof Burtseva EI, Fedorov SP and Zakharova EV; and to specialists of the DNA Sequencing Center of SB RAS (Novosibirsk): Morozov IV, Bondar AA and Tupikin AE for assistance in sequencing a part of our DNA samples. The study was supported by the Russian Foundation for Basic Research (#09-04-01123-a, #11-04-01221-a), Federal Programs ‘Scientific and educational staff for innovative Russia’ #02.740.11.0701 for years 2010–2012, #16.740.11.0190 and #16.740.11.0346 for years 2009–2013, the AI Ivanov Sakha Republic President Grant for Young Researchers in Medicine for 2009 year (03.02.2010 #71-p) and the State Scholarship of the Academy of Sciences of the Sakha Republic (Yakutia).
Ethics approval: This work was approved by the local bioethics committee at the Yakut Research Center of Complex Medical Problems of Siberian Branch of the Russian Academy of Medical Sciences (Yakutsk, Russian Federation). All participating families received written detailed information about this study. Written consent was obtained from all adult patients or from parents of infant patients.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Barashkov, N., Dzhemileva, L., Fedorova, S. et al. Autosomal recessive deafness 1A (DFNB1A) in Yakut population isolate in Eastern Siberia: extensive accumulation of the splice site mutation IVS1+1G>A in GJB2 gene as a result of founder effect. J Hum Genet 56, 631–639 (2011). https://doi.org/10.1038/jhg.2011.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.72
Keywords
This article is cited by
-
Genetic etiology of hearing loss in Russia
Human Genetics (2022)
-
A common founder effect of the splice site variant c.-23 + 1G > A in GJB2 gene causing autosomal recessive deafness 1A (DFNB1A) in Eurasia
Human Genetics (2022)
-
Autosomal recessive cataract (CTRCT18) in the Yakut population isolate of Eastern Siberia: a novel founder variant in the FYCO1 gene
European Journal of Human Genetics (2021)
-
Updated carrier rates for c.35delG (GJB2) associated with hearing loss in Russia and common c.35delG haplotypes in Siberia
BMC Medical Genetics (2018)
-
GJB2 c.−23+1G>A mutation is second most common mutation among Iranian individuals with autosomal recessive hearing loss
European Archives of Oto-Rhino-Laryngology (2015)
