
Abstract
Structural profiling of healthy human gut microbiota across heterogeneous populations is necessary for benchmarking and characterizing the potential ecosystem services provided by particular gut symbionts for maintaining the health of their hosts. Here we performed a large structural survey of fecal microbiota in 314 healthy young adults, covering 20 rural and urban cohorts from 7 ethnic groups living in 9 provinces throughout China. Canonical analysis of unweighted UniFrac principal coordinates clustered the subjects mainly by their ethnicities/geography and less so by lifestyles. Nine predominant genera, all of which are known to contain short-chain fatty acid producers, co-occurred in all individuals and collectively represented nearly half of the total sequences. Interestingly, species-level compositional profiles within these nine genera still discriminated the subjects according to their ethnicities/geography and lifestyles. Therefore, a phylogenetically diverse core of gut microbiota at the genus level may be commonly shared by distinctive healthy populations as functionally indispensable ecosystem service providers for the hosts.
Similar content being viewed by others

Introduction
The human gut microbiota is of great importance as indicated by accumulating evidence of its close association with human health (Clemente et al., 2012). As an internalized ‘microbial organ’, the gut microbiota provides many functions that are not encoded in the host genome but shown to be essential for human health, such as short-chain fatty acid (SCFA) production and vitamin synthesis (Flint et al., 2012, Gill et al., 2006). Such health-relevant functions may be considered ecosystem services provided by the gut microbiota for the host (Costello et al., 2012). The identification of specific phylotypes as key ecosystem service providers (ESPs) and the understanding of their interactions with each other and with their hosts are fundamental questions in human microbiome research. If these ESPs are indispensable to the host’s well being, healthy human individuals may share some key structural features, whereas diseased individuals may have aberrant patterns leading to a state of ‘dysbiosis’, which was found in obesity (Fei and Zhao, 2013), inflammatory bowel disease (Li et al., 2012), colorectal cancer (Wang et al., 2012) and type 2 diabetes (Qin et al., 2012) recently.
The focus on the common features of ‘normal’ gut microbiota in healthy human populations is increasing. It was reported that the human gut microbiota is typical of omnivorous primates (Ley et al., 2008). Thus far, the largest cohort study found that its composition and diversity varied widely even among healthy western individuals (Huttenhower et al., 2012). Particularly, ethnic background was strongly associated with a variety of microbial taxa, gene families and metabolic pathways. Also, variations were observed across geography, which reflected, at least partly, the differences in lifestyles and long-term diets between the cohorts (De Filippo et al., 2010, Yatsunenko et al., 2012). Age is another very important factor in shaping the gut microbiota, as exemplified by the dominance of Bifidobacterium in infants (Yatsunenko et al., 2012) and a greater proportion of Bacteroides in the elderly (Claesson et al., 2011).
Partly due to the large inter-individual variations driven by multiple factors, it was initially supposed that human individuals may not have a commonly shared, substantial phylogenetic core of gut microbiota (for example, overall abundance >0.5%) (Turnbaugh et al., 2009), although a functional core has been well described (Huttenhower et al., 2012, Qin et al., 2010, Turnbaugh et al., 2009). However, recent large cohort studies by deep, high-throughput sequencing found that within some genera, such as Bacteroides and Faecalibacterium, specific lineages were prevalent in at least 90% of the investigated western subjects (Huse et al., 2012, Qin et al., 2010, Zupancic et al., 2012). Alternatively, the human gut microbiota has also been classified into three compositional categories (known as enterotypes), primarily based on the dominance of the genera Bacteroides, Prevotella or Ruminococcus, which appeared independent of geography, sex, age and body mass index (Arumugam et al., 2011), but may be associated with long-term diets (Wu et al., 2011). Given the large variation in host properties and the habitat-dependent environmental factors across distinct human populations, further studies are needed for delineating the commonality of the proposed prevalent phylotypes and their contribution to human health.
To assess the impact of potential factors such as geography, lifestyle and ethnicity on shaping the gut microbiota, and to determine whether healthy adults can share common phylogenetic features in the context of these heterogeneities, we recruited 314 healthy young Chinese volunteers aged between 18 and 35 years, an age range regarded as the climax of human development in terms of vigor and health (Zastrow and Kirst-Ashman, 2012). These subjects belong to the widely dispersed Han people and six minority ethnic groups resident in their representative geographical locations in nine provinces throughout China, with both rural and urban lifestyles at each region, thereby representing healthy cohorts with highly diverse genetic, cultural and dietary backgrounds. By combining deep 454 pyrosequencing (Margulies et al., 2005) and multivariate statistics, we uncovered nine predominant genus-level taxa known to contain SCFA producers, which co-occurred in all individuals and may represent a phylogenetic core as potential ESPs beneficial to human health.
Materials and methods
Subject selection and sampling
This study was approved by the Inner Mongolia Agriculture University, and informed consent was obtained from all the 314 healthy volunteers before enrollment in the study. All volunteers (body mass index 18.4–25.7 kg m−2) recruited in this study were indigenous residents and had resided in the same locality for at least three generations without marrying with individuals from any other ethnic groups. The identity and ethnicity of each volunteer was confirmed by checking his/her household register, and the consistency of his/her language, dressing style and lifestyle with that of the corresponding ethnic group. In each sampling site, paired volunteers who were matched/nearly matched for sex and urban/rural lifestyle were recruited. None of these volunteers had gastrointestinal tract disorders or taken any antibiotics for at least 3 months prior to the sampling. Fresh fecal samples were collected in the early morning before breakfast under anaerobic conditions and frozen immediately in liquid nitrogen. Samples were transferred to the laboratory within 24 h in an ice bucket and stored at −80 °C.
Pyrosequencing, sequence preprocessing, and bioinformatics
DNA extraction and PCR amplification of the V5–V6 region of 16S rRNA genes were performed as described in Supplementary Information and previous studies (Dethlefsen and Relman, 2011, Lee et al., 2011). PCR products from different samples were mixed at equal ratios for pyrosequencing with the GS FLX platform (Roche, Branford, CT, USA; University of Hawaii, Honolulu, HI, USA) and a total of 13 runs were performed for the 314 samples. Samples from at least three ethnic groups were blended in each run, to ensure that there was no case that samples from one single ethnic group were sequenced in a single run (Supplementary Table S2).
Operational taxonomic unit (OTU) classification, chimera removal, tree construction and taxonomic assignment were all performed using the QIIME package (Quantitative Insights Into Microbial Ecology, Boulder, CO, USA, v1.2.1) (Caporaso et al., 2010) after removing the bad-quality sequences from the raw data set (Supplementary Information). To minimize the difference of sequencing depth among samples, the rarefied OTU subset was generated by averaging 1000 evenly resampled OTU subsets for further alpha and beta diversity analysis (Supplementary Information).
Statistical analysis
Statistical analysis was conducted mainly under Matlab environment (The MathWorks, Natick, MA, USA) and R (http://www.r-project.org/). Based on the rarefied OTU subset, the relative abundances of taxa were compared among cohorts by the Kruskal–Wallis test. Spearman’s rank correlation was used to evaluate the co-occurrence relationships between the relative abundances of taxa. False discovery rate (FDR) values were estimated using the Benjamini–Yekutieli method (Benjamini and Yekutieli, 2001) to control for multiple testing. Canonical analysis of principal coordinates (CAP) (Anderson and Willis, 2003) and permutational multivariate analysis of variance (PERMANOVA) (McArdle and Anderson, 2001) were employed to evaluate if the gut microbiota structure was significantly segregated across the cohorts. CAP was performed using the FORTRAN program (Anderson, 2004) released by the authors proposing the CAP method (Anderson and Willis, 2003) and PERMANOVA was performed using the function ‘adonis’ implemented in the R ‘vegan’ package (Oksanen et al., 2013). For both statistical methods, the UniFrac distance matrix was used as the input of the microbial community data, whereas the ethnicity, geography, sex or lifestyle of each subject was used as the input of the factor (that is, the constraint). After the principal coordinates analysis (PCoA) in CAP, the first m principal coordinates were determined as the optimal dimensionality by (1) estimating the minimal misclassification error yielded by the leave-one-out cross-validation; and (2) computing the proportion of the total variations in the original distance matrix explained by the first m principal coordinates, which must exceed 60% but must not exceed 100% (Anderson and Willis, 2003). The canonical plot was generated based on the sample scores of the first three canonical axes. The significance of the results from CAP and PERMANOVA was tested by 9999 permutations. Enterotype analysis based on the genus-level abundance profiles was performed according to the methods proposed in the original report of enterotypes (Arumugam et al., 2011) using the R ‘ade4’ package.
Results
Study cohorts
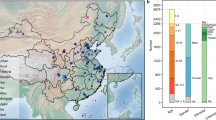
We recruited 314 healthy, young, unrelated volunteers, including 145 urban and 169 rural residents, with an overall equivalent sex ratio and representing 20 cohorts from 7 ethnic groups (that is, the Bai, Han, Kazakh, Mongol, Tibetan, Uyghur and Zhuang) from 9 provinces in China (Figure 1, Supplementary Tables S1 and S2). We recruited the Han volunteers from four representative geographical locations in Northeast (Harbin), Central (Zhengzhou), East (Wuxi) and Southwest (Chengdu) China with distinct natural environmental conditions and economic status. Each of the other six non-Han minority ethnic groups was recruited from a single region of their representative locations, respectively. Particularly, the Kazakh and Uyghur resided in similar habitats. All the recruited rural residents lived far away from the metropolitan areas with typical farming or pastoral lifestyle.

Cohorts investigated in this study. Sampling sites are mapped using Esri ArcMap 10.1. The sampling sites of the urban Kazakh and Uyghur ethnic groups are both located in Ürümqi and thus are overlapped on this physical map. Scale bar: 1000 km.
Diversity of gut microbiota in healthy young Chinese cohorts
A total of 5 102 015 high-quality sequences were generated by pyrosequencing the V5–V6 region of the 16S ribosomal RNA (rRNA) gene of Bacteria and Archaea (Lee et al., 2011) from fecal samples collected from the 314 volunteers (median=13 361 sequences, ranging from 5992 to 96 710 sequences; Supplementary Table S2). At a threshold of 97% sequence identity, 36 918 OTUs were identified in the current study (median=1821 OTUs, ranging from 555 to 7724 OTUs; Supplementary Table S2), representing 4 406 917 sequences for subsequent analysis (Supplementary Information). Although no OTU-level rarefaction curves plateaued under the current sequencing depth (Supplementary Figures S1a–j), all the Shannon diversity indices reached stable values (Supplementary Figures S1k–t), suggesting that most of the microbial diversity had already been captured in this data set despite the possibility to detect rare new phylotypes with additional sequencing efforts.
Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria constituted the four most dominant bacterial phyla (contributing 73.47, 14.13, 5.83 and 3.36% of the total sequences, respectively), whereas Euryarchaeota was the only detected phylum in the domain of Archaea (2.62%). Consistent with previous studies in African, Asian and western cohorts (De Filippo et al., 2010, Huttenhower et al., 2012, Nam et al., 2011), Firmicutes and Bacteroidetes were the two most abundant phyla composing the vast majority of the gut microbiota of the healthy cohorts in this study, but the ratio of Firmicutes: Bacteroidetes had considerable inter-individual variations within each cohort (ranging from 0.5 to 300.6, Supplementary Figure S2a). Such wide variations also poorly linked with the body weight and the body mass index (P=0.75 and 0.97 by Spearman’s rank correlation, respectively; Supplementary Figures S2b and c). Only 51.60% of the OTUs (contributing to 77.42% of the total sequences) were assigned to known genera, indicating the potential to discover novel genus-level taxa by deeper sequencing. Among the 279 identified genera, Phascolarctobacterium of Firmicutes was the most predominant and more abundant than the generally recognized predominant taxa, such as Bacteroides (Arumugam et al., 2011) and Faecalibacterium (Nam et al., 2011); it also showed the most variable abundance across all individuals (Supplementary Table S3). Of the 16 most abundant genera, 11 were Firmicutes (Supplementary Table S3).
Within the domain of Archaea, it was reported that Methanobrevibacter smithii could comprise up to 11.4% of the whole gut microbiota (Eckburg et al., 2005). Besides M. smithii, Methanosphaera stadtmanae is also sometimes detectable in human gut microbiota, with lower and varying abundance (Dridi et al., 2009, Hoffmann et al., 2013, Lecours et al., 2014). In this study, Euryarchaeota was dominated by Methanobrevibacter (2.09%) and Methanosphaera (0.44%). Methanobrevibacter was identified in 259 samples, yielding a similar prevalence (82.48%) compared with previous study (Dridi et al., 2009). The prevalence of Methanosphaera (57.32%) was relatively higher than that reported previously (Dridi et al., 2009, Hoffmann et al., 2013, Lecours et al., 2014). Both genera were absent in 41 individuals and represented >10% of the whole gut microbial community in 23 and 3 individuals, respectively. The abundance of both genera varied largely across individuals (ranging from 0% to 39.10% for Methanobrevibacter and ranging from 0% to 17.72% for Methanosphaera). These two genera varied significantly among ethnic groups (Supplementary Table S4) and tended to be enriched in rural individuals (Supplementary Table S5), indicating that their prevalence and abundance may vary with the particular populations studied.
Structural segregation of gut microbiota across cohorts
To profile the overall gut microbiota structure, a set of unconstrained ordination methods were applied to the rarified OTU data (4561 sequences per sample). Large inter-individual compositional variation was observed through principal component analysis (Supplementary Figure S3a) and PCoA based on the weighted UniFrac distance (Supplementary Figure S3b) (Lozupone et al., 2007). Unweighted UniFrac PCoA (Lozupone and Knight, 2005) well separated the Mongol from the Tibetan ethnic group (Figure 2a and Supplementary Figure S3c); the differentiation between rural and urban cohorts was also evident within the Mongol and Zhuang ethnic groups (Supplementary Figures S3c and d).

The healthy young Chinese cohorts show significant ethnicity-associated structural segregation of gut microbiota. (a) Unweighted UniFrac PCoA of the 314 samples using the full set of OTUs. The percentage of the variation explained by the plotted principal coordinates (PCs) is shown in parentheses. (b) CAP using the first 198 PCs. The squared canonical correlation values (δ2) are shown in parentheses. Permutation tests reveal that the segregation is significant (P=0.0001).
To maximally extract the information derived from unweighted UniFrac PCoA and better understand the impact of lifestyle and ethnicity on the gut microbiota structure, we introduced CAP (Anderson and Willis, 2003), a constrained ordination method friendly to any kind of distance metrics. Using the first 198 principal coordinates (accounting for 82.85% of the total variation), 223 individuals were successfully predicted by their ethnicities, yielding a cross-validated correct classification rate (CCR) of 71.02% as well as the highly significant canonical test statistics (P=0.0001). As expected, the canonical plot (Figure 2b) showed high squared canonical correlations (δ2) and a more distinct segregation among ethnic groups compared with the original UniFrac PCoA plot (Figure 2a) over the first three dimensions. The Tibetan group was entirely separated from the others, forming an ‘out-group’ with the highest CCR (86.05%); the Mongol and Zhuang were also wide apart from each other (CCR=77.08% and 71.74%, respectively). Within the Han ethnic group from diverse and distant habitats, 77 out of the 91 individuals (84.62%) were correctly classified. Twenty-five out of the 43 Bai individuals were successfully classified and the others were misclassified as Han (CCR=58.14%), resulting in a partial overlap between these two ethnic groups in the canonical plot (Figure 2b). The Kazakh and Uyghur, the two ethnic groups sharing similar milieus in Xinjiang Uyghur Autonomous Region (all the recruited urban individuals of both groups were residents of Ürümqi) and similar ethnic origin (both are the Turkic descendants), were not well distinguishable from each other (CCR=22.73% and 42.86%, respectively). Such significant structural segregation of the gut microbiota across ethnic groups was also confirmed by PERMANOVA (P=0.0001), a method that can assess the effects of factors directly based on unweighted UniFrac or other distance matrices (McArdle and Anderson, 2001). Particularly, for the Han cohorts, the gut microbiota structure was significantly segregated across the four geographical regions (P=0.0001). Additionally, other potential factors, such as sex, showed a trend but not significant in the segregation of the gut microbiota structure across all the individuals (P=0.0531).
Structural differentiation of gut microbiota between the rural and urban cohorts varied greatly across geographical regions and ethnic groups (Table 1). The Mongol cohorts exhibited the highest predictive power of gut microbiota structure between lifestyles among all the ethnic groups (CCR=97.92%). The Mongolians also had the largest lifestyle-associated difference in Shannon diversity indices (8.66±1.39 vs 6.74±0.95, P<10−5 by Student’s t-test). Interestingly, the Han living in different geographical regions showed a wide range of CCR, from 23.5% to 73.91%. Similarly, PERMANOVA revealed that the impact of lifestyle was most significant within the Mongol and Zhuang ethnic groups, followed by the Han cohorts living in Harbin and Zhengzhou, whereas this impact was marginally significant in the Tibetan cohorts (see Table 1 for the corresponding P-values). In contrast, the Uyghur and Kazakh showed the smallest lifestyle-associated distances (Supplementary Figure S4). Within the Han ethnic group, the individuals living in Chengdu and Wuxi had significantly smaller distance values than those in Zhengzhou and Harbin, which were in agreement with the results of CAP and PERMANOVA.
A number of genera were differentiated between the rural and urban individuals (Supplementary Tables S5 and S6). For example, Prevotella and Xylanibacter of Bacteroidetes were among the taxa most significantly enriched in all the investigated rural individuals, while Lactobacillus, Methanobrevibacter, Methanosphaera and Methanobacterium were particularly more abundant in the rural Mongol individuals than in their urban counterparts.
We further defined the most similar and dissimilar ‘neighbors’ to each cohort in terms of their gut microbiota structure, by comparing the cohort-to-cohort average UniFrac distances (Supplementary Figure S5). The rural Tibetan cohort was identified as the most dissimilar one to all the other cohorts except the rural Uyghur (to which the most dissimilar cohort was the rural Han living in Zhengzhou), again highlighting the large structural discrepancy of gut microbiota between the Tibetan and other ethnic groups. Although the rural Uyghur was the most similar one to the rural and urban Tibetan cohorts, it still showed relatively high distance values (0.7330±0.0029 and 0.7220±0.0023, respectively). The smallest cohort-to-cohort distance value was between the rural Mongol and the urban Uyghur cohort (0.6638±0.0024); the latter was also the most similar cohort to its rural counterpart.
A genus-level phylogenetic core shared by all the subjects
We tested whether any genus- or OTU-level phylotypes occurred in each investigated individual to compose a core gut microbiota (Hamady and Knight, 2009). Nine predominant genera were present in all the investigated individuals (Supplementary Table S3), constituting a genus-level phylogenetic core accounting for 47.63% of the total sequences (Figure 3a). All the genera in this core were Firmicutes except Bacteroides, and all of these taxa occupied >1% of the individual gut microbial community in at least 75% of all subjects except Coprococcus (in 69.11% subjects) (Figure 3b). The collective core was overwhelmingly dominant (abundance >50%) in more than half the subjects (50.96%) and occupied <1% of the whole gut microbial community in only one individual (Figure 3b) but showed dramatic variations in abundance in each investigated cohort, regardless of geography, lifestyle and ethnicity (Figure 3c).

The cohorts share a core gut microbiota composed of nine bacterial genera. (a) The proportion of each genus in the total sequences. (b) The abundance distribution of the nine genera and the collective core. Boxes represent the interquartile range (IQR) between the first and third quartiles. The lines and squares inside boxes represent the median and mean, respectively. Whiskers denote the lowest and highest values within 1.5 × IQR from the first and third quartiles, respectively. (c) The abundance of the collective core varies greatly across individuals in each cohort. Individuals in each cohort are arranged according to their abundance of the collective core. The cohorts are symbolized as in Figure 1.
Spearman’s rank correlation test was employed to identify the co-occurrence patterns among the nine core genera (Figure 4). Besides the genus Phascolarctobacterium, which only had a very weak co-exclusion relationship with Clostridium (rho=−0.195, FDR=0.30%), the other eight core genera were positively correlated with each other (Figure 4). This was most significant between the two phylogenetically related genera Faecalibacterium and Subdoligranulum (rho=0.664, FDR=0%) and also among Clostridium, Blautia and Ruminococcus (rho>0.5, FDR=0%).

Co-occurrence patterns among the nine core genera across the 314 samples, as determined by the Spearman’s rank correlation analysis. The correlations were controlled for multiple testing and only those with FDRs <1% were retained. The significant correlations were subsequently converted to correlation distance matrices and the taxa were clustered using the UPGMA (Unweighted Pair Group Method with Arithmetic Mean) hierarchical clustering method. *FDR<0.01, **FDR<0.001 and ***FDR<10−5.
At the genus level, we did not identify the enterotypes reported in western cohorts mainly driven by the predominant genera Bacteroides, Prevotella and Ruminococcus (Arumugam et al., 2011). Although partitioning around medoids analysis revealed that the optimal number of clusters was two, such clustering could not be well supported by reliable silhouette width values (Supplementary Figure S6a) and the clusters nearly overlapped in between-class analysis (Supplementary Figure S6b).
At the OTU level, none was detectable in all individuals; most OTUs were shared by only a limited number of individuals (Supplementary Figure S7). Instead, we found 30 OTUs shared by at least 90% of all the individuals (Supplementary Table S7). They represented the most prevalent OTU-level phylotypes in this study, corresponding to only a minor part of all the 36 918 OTUs (0.081%) but 838 125 of all the 4 406 917 sequences (19.018%). All these OTUs were Firmicutes except OTU61903 (belonging to Collinsella from Actinobacteria) and OTU75613 (belonging to Klebsiella from Proteobacteria). The most prevalent OTU (OTU70817) belonged to Faecalibacterium, which was detected in 306 out of the 314 individuals (97.5%).
Significantly positive co-occurrence relationships among the 30 most prevalent OTUs were not only widely identified among the distantly related OTUs, such as between OTU61903 (belonging to Collinsella) and OTU89633 (Dorea) (rho=0.375, FDR=1.80 × 10−10) and between OTU8392 (Phascolarctobacterium) and OTU75613 (Klebsiella) (rho=0.287, FDR=3.93 × 10−06), but also among the OTUs with close phylogenetic relationships, exemplified by the three OTUs within Blautia (rho values ranged between 0.535 and 0.679, FDR ⩽9.02 × 10−23), and the four OTUs within Dorea (rho values ranged between 0.514 and 0.766, FDR ⩽9.77 × 10−21). The highest positive correlation was found between OTU43582 (Coprococcus) and OTU69334 (currently unclassified Firmicutes) (rho=0.907, FDR=7.12 × 10−117) (Supplementary Figure S8).
Species-level segregation within the core genera
To test whether species-level composition within the core gut microbiota could be still segregated by lifestyle and ethnicity/geography, we applied the same set of ordination methods described above to the 10 937 OTUs assigned to the nine core genera. Unweighted UniFrac PCoA revealed a similar sample distribution to that based on the full set of OTUs, particularly so in the three most distinctive ethnic groups, the Mongol, Tibetan and Zhuang (Figure 5a and Supplementary Figure S9). Using the first 176 principal coordinates (accounting for 84.20% of the total variation), CAP successfully predicted 218 individuals by their ethnicities (CCR=69.43%), almost as well as that obtained from the full model. The canonical test statistics were also highly significant (P=0.0001) and the first three δ2 were still high (Figure 5b). The clustering pattern in the canonical plot (Figure 5b) was also consistent with that derived from the full set of OTUs (Figure 2b). The Tibetan ethnic group still had the highest CCR (83.72%) and was, again, entirely separated from others, followed by the Han, Mongol, Zhuang and Bai cohorts (CCR=80.22, 77.08, 63.04 and 60.47%, respectively). The Kazaka and Uyghur ethnic groups were still largely misclassified (CCR=31.82% and 47.62%, respectively). This structural segregation across ethnic groups was also confirmed by PERMANOVA (P=0.0001), as well as the segregation across geography in the Han cohorts (P=0.0007). The lifestyle-associated differentiation was also consistent with that based on the full set of OTUs, without marked changes in the statistics obtained by the CAP procedure and PERMANOVA (Table 1).

The genus-level core gut microbiota shows significant ethnicity-associated structural segregation at the species level. (a) Unweighted UniFrac PCoA of the 314 samples using the 10,937 OTUs assigned to the nine core genera. The percentage of the variation explained by the plotted PCs is shown in parentheses. (b) CAP using the first 176 PCs. δ2 Values are shown in parentheses. Permutation tests reveal that the segregation is significant (P=0.0001).
Discussion
In anthropology, an ethnic group is ‘a named social group based on perceptions of shared ancestry, cultural traditions and common history that culturally distinguish that group from other groups’ (Peoples and Bailey, 2011). As a vast country, China occupies nearly 20% of the world population, including the wide-ranging Han and the other 55 minority ethnic groups living in their local habitats, which have endemic characteristics in genetics (Chu et al., 1998), lifestyle, diet, culture and so on. Given the tight relationship between the gut microbiota and these factors (De Filippo et al., 2010, Huttenhower et al., 2012, Tyakht et al., 2013, Yatsunenko et al., 2012), the Chinese people with truly diverse ethnic origins offer us good opportunities to visit and understand the diversity, variability and commonality of gut microbiota. In this study, the significant, multi-factor-associated structural segregation of the gut microbiota across the cohorts was unraveled by combining the unsupervised and constrained multivariate analytic tools on the basis of unweighted (a qualitative measure) (Lozupone and Knight, 2005) rather than weighted UniFrac distance (a quantitative measure) (Lozupone et al., 2007), which indicated that the gut microbiota structure of the investigated cohorts was differentiated primarily by the microbial lineages surviving on the host’s specific ethnicity but not the variation in their abundance. Such segregation was particularly significant on the Mongol and Tibetan ethnic groups, which coincided with the fact that both of them usually live as typical nomadic tribes (for example, hunter–gatherers and cattle herders) on the Xilingol Grassland and Qinghai-Tibetan Plateau, respectively. Although most rural and urban Han individuals were clustered together in the same group in Figure 2b, the Han cohorts recruited from the four geographical regions harbored significantly distinctive gut microbiota structure, suggesting that geography may also exert selection pressure on the human gut microbiota. As the habitats of most minority ethnic groups are much more specific than that of the Han, the impact of ethnicity on the gut microbiota structure observed in this study requires deeper investigation to understand its complex and profound interactions with other confounding factors, such as geography. This objective can be achieved by comparing the gut microbiota of these minorities with that of their local Han counterparts.
Lifestyle was another major factor in the structural segregation of gut microbiota, but its effect varied across geography and ethnicity, particularly for the Han cohorts (Supplementary Information). The Mongol ethnic group represented the most significant structural differentiation between the rural and urban cohorts in this study. Generally, the rural Mongol keeps the nomadic lifestyle, lives far away from metropolitan areas like Hohhot, relies on their traditional fermented dairy products and spends substantial amount of their lifetime looking for pastures and campsites, whereas their urban counterparts no longer need to strictly follow such tradition and have adapted to the modern lifestyle, including the urbanized diet. These large differences in lifestyle and long-term dietary patterns may influence their gut microbiota profoundly. For example, Lactobacillus, one genus frequently isolated from Mongolian fermented dairy products (Wu et al., 2009), was more abundant in the rural Mongol cohort. The global enrichment of Prevotella and Xylanibacter in all the investigated rural cohorts may represent another notable structural feature of the gut microbiota. These two taxa were reported to be enriched in rural Africa children but under-represented in the European ones; the prominence of such carbohydrate-utilizing, SCFA-producing bacteria with elevated fecal SCFA concentrations may promote the energy intake from fibers, inhibit opportunistic pathogens and protect the hosts against inflammation and colonic diseases (De Filippo et al., 2010). High level of Prevotella was also associated with the carbohydrate-based diet, a typical characteristic of rural populations and agrarian societies, rather than the ‘western’ diet rich in animal protein and saturated fats (Wu et al., 2011).
Despite the wide structural variations across ethnicity/geography and lifestyle at the OTU level, we still characterized a phylogenetic core of gut microbiota prevalent in unrelated individuals, which was made up of a small number of genus-level phylotypes. Most of these phylotypes were also the most prevalent ones in the western population (Zupancic et al., 2012). Interestingly, this supposed phylogenetic core showed potential for functional commonality: All the nine core genera contain known SCFA-producing lineages. For example, Bacteroides mainly produces succinate and acetate (Shah and Collins, 1989); Phascolarctobacterium produces propionate via succinate fermentation (Dot et al., 1993) (See Supplementary Information for its predominance and co-occurrence relationships with other bacterium); Blautia is known as the acetogen (Park et al., 2012); Roseburia, Faecalibacterium and Subdoligranulum are butyrate producers (Duncan et al., 2002a, Duncan et al., 2002b, Holmstrom et al., 2004). SCFAs, primarily acetate, propionate and butyrate, are major anions in the gut and are absorbed rapidly by colonic epithelial cells. A substantial amount of acetate enters systemic circulation and is used principally by peripheral tissues for lipogenesis; propionate is largely consumed by the liver for gluconeogenesis; butyrate is the preferred energy source for colonocytes (Pomare et al., 1985, Scott et al., 2008). Accumulating evidence indicates that these small molecules can improve the gut barrier function (Peng et al., 2009), suppress insulin-mediated fat accumulation in adipose tissue (Kimura et al., 2013), exhibit anti-inflammatory effects (Maslowski et al., 2009, Segain et al., 2000) and protect the host against colonic diseases (Fukuda et al., 2011, McIntyre et al., 1993). Depletion of some universal butyrate-producers along with enrichment of various opportunistic pathogens in the gut microbiota was identified in colorectal cancer (Wang et al., 2012) and type 2 diabetes (Qin et al., 2012) by recent microbiome-wide association studies. Particularly, Faecalibacterium prausnitzii, which produces high amounts of butyrate and exhibits anti-inflammatory effects, has shown potential as a probiotic in the treatment of Crohn’s disease (Sokol et al., 2008) and hence may be considered as a crucial ESP for human health (Costello et al., 2012). Lineages within Faecalibacterium were commonly shared by healthy western individuals described previously (Huse et al., 2012, Qin et al., 2010, Tap et al., 2009, Zupancic et al., 2012). Here Faecalibacterium was one of the core members and represented the most prevalent OTU. The prevalence of such lineages across heterogeneous populations may indicate their essentiality for human health.
SCFA production may represent a pivotal feature of the core consisting of the nine genera. Other predominant SCFA-producing genera such as Prevotella (Shah and Collins, 1990) and Dorea (Taras et al., 2002) was also detectable in >97% of all individuals at current sequencing depth and hence may also be candidates for the phylogenetic core. The acetate-producing Bifidobacterium, however, showed a lower prevalence, indicating the probability that this genus may not serve as a fundamental member of the most commonly shared core in healthy young individuals, although it is considered as an major beneficial symbiont in human gastrointestinal tract (Fukuda et al., 2011).
In addition to the presumed capacity for SCFA production, our work also implied that the core genera were more likely to co-occur, even between the phylogenetically similar taxa (for example, Faecalibacterium and Subdoligranulum), rather than to competitively exclude each other. Such co-occurrence relationships were even more significant among the prevalent OTUs belonging to Blautia or Dorea. In contrast to the expectation that closely related lineages could compete against each other due to their overlapping habitats and ecological roles, it was found that such lineages tend to co-occur within the same niche (Faust et al., 2012). Taxon pairs with >98.5% identity were also found in the same co-occurrence module constantly (Lozupone et al., 2012). As the core genera characterized here might be involved in the metabolic network of various SCFAs, converge on some specific biological attributes and share similar environmental preferences, these findings substantiated the high levels of functional redundancy in the core gut microbiota across healthy young human individuals. Within the core, each member might also avoid competition with others and function as specific ESPs for the host by utilizing particular resource as substrates (for example, a specific kind of carbohydrates such as cellulose, resistant starch and oligosaccharides) to produce specific metabolic profiles (for example, a few kinds of SCFAs), which may in turn induce these taxa to be selected in the host simultaneously. For example, Blautia schinkii ferments D-arabinose but not L-arabinose, which can be utilized by Blautia stercoris (Park et al., 2012).
Another intriguing feature of the phylogenetic core described here was its significant species-level variability across the cohorts. The same set of core genera may be universally retained in unrelated healthy individuals to provide some conserved ecosystem services to maintain human health, but the exact composition of the core gut microbiota at the species and finer (strain) levels may, most likely, coevolve with their hosts due to selection pressures, such as lifestyle and ethnicity/geography. The mutualistic interactions between the hosts and the symbiotic gut microbiota could be inherited and selected to confer essential benefits to the hosts, through vertical transmission of whole gut microbial communities from parents to offspring (Ley et al., 2006), which would lead to the evolution of a cohort-specific, and probably health-associated, core gut microbiota structure.
Taken together, despite the large structural segregation of gut microbiota due to the diverse ethnicities/geography and lifestyles of the healthy cohorts studied here, we defined a phylo-functional core of gut microbiota, that is, the assemblage of a few bacterial genera with potentially conserved but indispensable functions for human health, such as the SCFA production. The establishment of such a phylo-functional core across healthy populations may serve as a benchmark for linking the gut microbiota structure with the crucial health-associated ecosystem services that it provides to their hosts. Targeted nourishment of these beneficial, core symbionts may thus provide a promising strategy in the improvement of human health. Further studies are required to unveil the functional roles conserved in these core phylotypes, to exploit their interactions with each other, with various pathogens as well as with their hosts, and to focus on the ecological and evolutionary principles of the core for maintaining the homeostasis of gut ecosystem and thus on human health.
Sequence data accession number
All sequence data have been deposited in the MG-RAST database 3.3 (accession no. 1538).
Accession codes
References
Anderson MJ, Willis TJ . (2003). Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84: 511–525.
Anderson MJ . (2004). CAP: a FORTRAN computer program for canonical analysis of principal coordinates. Ecological Archives E084-011-S1: Ithaca, New York, USA.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al. (2011). Enterotypes of the human gut microbiome. Nature 473: 174–180.
Benjamini Y, Yekutieli D . (2001). The control of the false discovery rate in multiple testing under dependency. Ann Stat 29: 1165–1188.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E et al. (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 108: 4586–4591.
Clemente JC, Ursell LK, Parfrey LW, Knight R . (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270.
Costello EK, Stagaman K, Dethlefsen L, Bohannan BJM, Relman DA . (2012). The application of ecological theory toward an understanding of the human microbiome. Science 336: 1255–1262.
Chu JY, Huang W, Kuang SQ, Wang JM, Xu JJ, Chu ZT et al. (1998). Genetic relationship of populations in China. Proc Natl Acad Sci USA 95: 11763–11768.
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA 107: 14691–14696.
Dethlefsen L, Relman DA . (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA 108: 4554–4561.
Dot TD, Osawa R, Stackebrandt E . (1993). Phascolarctobacterium faecium gen. nov, spec. nov., a novel taxon of the Sporomusa group of Bacteria. Syst Appl Microbiol 16: 380–384.
Dridi B, Henry M, El Khechine A, Raoult D, Drancourt M . (2009). High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS One 4: e7063.
Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ . (2002a). Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int J Syst Evol Microbiol 52: 1615–1620.
Duncan SH, Hold GL, Harmsen HJM, Stewart CS, Flint HJ . (2002b). Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int J Syst Evol Microbiol 52: 2141–2146.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al. (2005). Diversity of the human intestinal microbial flora. Science 308: 1635–1638.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8: e1002606.
Fei N, Zhao LP . (2013). An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J 7: 880–884.
Flint HJ, Scott KP, Louis P, Duncan SH . (2012). The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 9: 577–589.
Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K et al. (2011). Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469: 543–547.
Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312: 1355–1359.
Hamady M, Knight R . (2009). Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res 19: 1141–1152.
Hoffmann C, Dollive S, Grunberg S, Chen J, Li HZ, Wu GD et al. (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8: e66019.
Holmstrom K, Collins MD, Moller T, Falsen E, Lawson PA . (2004). Subdoligranulum variable gen. nov., sp. nov. from human feces. Anaerobe 10: 197–203.
Huse SM, Ye YZ, Zhou YJ, Fodor AA . (2012). A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7: e34242.
Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT et al. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214.
Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T et al. (2013). The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun 4: 1829.
Lecours PB, Marsolais D, Cormier Y, Berberi M, Hache C, Bourdages R et al. (2014). Increased prevalence of Methanosphaera stadtmanae in inflammatory bowel diseases. PLoS One 9: e87734.
Lee OO, Wang Y, Yang JK, Lafi FF, Al-Suwailem A, Qian PY . (2011). Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J 5: 650–664.
Ley RE, Peterson DA, Gordon JI . (2006). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124: 837–848.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. (2008). Evolution of mammals and their gut microbes. Science 320: 1647–1651.
Li E, Hamm CM, Gulati AS, Sartor RB, Chen HY, Wu X et al. (2012). Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS One 7: e26284.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Lozupone C, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J et al. (2012). Identifying genomic and metabolic features that can underline early successional and opportunistic lifestyles of human gut symbionts. Genome Res 22: 1974–1984.
Lozupone CA, Hamady M, Kelley ST, Knight R . (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73: 1576–1585.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286.
McArdle BH, Anderson MJ . (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82: 290–297.
McIntyre A, Gibson PR, Young GP . (1993). Butyrate production from dietary fibre and protection against large bowel cancer in a rat model. Gut 34: 386–391.
Nam YD, Jung MJ, Roh SW, Kim MS, Bae JW . (2011). Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS One 6: e22109.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB et al. (2013). vegan: community ecology package. R package version 2.1-26/r2441.
Park SK, Kim MS, Roh SW, Bae JW . (2012). Blautia stercoris sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 62: 776–779.
Peng LY, Li ZR, Green RS, Holzman IR, Lin J . (2009). Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr 139: 1619–1625.
Peoples J, Bailey G . (2011). Ethnicity and ethnic conflict. In: Mitchell E (ed) Humanity: An Introduction to Cultural Anthropology 9th edn. Wadsworth, Cengage Learning: Belmont, California, USA, p 389.
Pomare EW, Branch WJ, Cummings JH . (1985). Carbohydrate fermentation in the human colon and its relation to acetate concentrations in venous blood. J Clin Invest 75: 1448–1454.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490: 55–60.
Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65.
Scott KP, Duncan SH, Flint HJ . (2008). Dietary fibre and the gut microbiota. Nutr Bull 33: 201–211.
Segain JP, de la Bletiere DR, Bourreille A, Leray V, Gervois N, Rosales C et al. (2000). Butyrate inhibits inflammatory responses through NFκB inhibition: implications for Crohn's disease. Gut 47: 397–403.
Shah HN, Collins MD . (1989). Proposal to restrict the genus Bacteroides (Castellani and Chalmers) to Bacteroides fragilis and closely related species. Int J Syst Bacteriol 39: 85–87.
Shah HN, Collins DM . (1990). Prevotella, a new genus to include Bacteroides melaninogenicus and related species formerly classified in the genus Bacteroides. Int J Syst Bacteriol 40: 205–208.
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ et al. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105: 16731–16736.
Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP et al. (2009). Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 11: 2574–2584.
Taras D, Simmering R, Collins MD, Lawson PA, Blaut M . (2002). Reclassification of Eubacterium formicigenerans Holdeman and Moore 1974 as Dorea formicigenerans gen. nov., comb. nov., and description of Dorea longicatena sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 52: 423–428.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Tyakht AV, Kostryukova ES, Popenko AS, Belenikin MS, Pavlenko AV, Larin AK et al. (2013). Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun 4: 2469.
Wang TT, Cai GX, Qiu YP, Fei N, Zhang MH, Pang XY et al. (2012). Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J 6: 320–329.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 333: 105–108.
Wu R, Wang LP, Wang JC, Li HP, Menghe B, Wu JR et al. (2009). Isolation and preliminary probiotic selection of lactobacilli from koumiss in Inner Mongolia. J Basic Microbiol 49: 318–326.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al. (2012). Human gut microbiome viewed across age and geography. Nature 486: 222–227.
Zastrow CH, Kirst-Ashman KK . (2012). Biological aspects of young and middle adulthood. In: Dobrin S (ed) Brooks/Cole Empowerment Series: Understanding Human Behavior and the Social Environment 9th edn. Brooks/Cole, Cengage Learning: Belmont, California, USA, pp 440–466.
Zupancic ML, Cantarel BL, Liu Z, Drabek EF, Ryan KA, Cirimotich S et al. (2012). Analysis of the gut microbiota in the old order Amish and its relation to the metabolic syndrome. PLoS One 7: e43052.
Acknowledgements
We appreciate the amendments from Professor Yuan Kun Lee of National University of Singapore. This work is supported by funds from the National Natural Science Foundation of China (Grant nos. 31025019 and 31125021), the Innovation Team Development of the Ministry of Education of China (Grant no. IRT0967) and the China Agriculture Research System (Grant no. CARS-37).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zhang, J., Guo, Z., Xue, Z. et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J 9, 1979–1990 (2015). https://doi.org/10.1038/ismej.2015.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.11
This article is cited by
-
Non-alcoholic fatty liver disease in patients with morbid obesity: the gut microbiota axis as a potential pathophysiology mechanism
Journal of Gastroenterology (2024)
-
Gut microbiota characteristics of Mongolian and Han populations in anti-tuberculosis drug-induced liver injury: a population-based case–control study
BMC Microbiology (2023)
-
Lifestyle patterns influence the composition of the gut microbiome in a healthy Chinese population
Scientific Reports (2023)
-
A display and analysis platform for gut microbiomes of minority people and phenotypic data in China
Scientific Reports (2023)
-
Exploring the influences of geographical variation on sequence signatures in the human gut microbiome
Journal of Genetics (2023)
