
Abstract
Hypoliths (cryptic microbial assemblages that develop on the undersides of translucent rocks) are significant contributors to regional C and N budgets in both hot and cold deserts. Previous studies in the Dry Valleys of Eastern Antarctica have reported three morphologically distinct hypolithic community types: cyanobacteria dominated (type I), fungus dominated (type II) and moss dominated (type III). Here we present terminal-restriction fragment length polymorphism analyses to elucidate the bacterial community structure in hypolithons and the surrounding soils. We show clear and robust distinction in bacterial composition between bulk surface soils and hypolithons. Moreover, the bacterial assemblages were similar in types II and III hypolithons and clearly distinct from those found in type I. Through 16S rRNA gene 454 pyrosequencing, we show that Proteobacteria dominated all three types of hypolithic communities. As expected, Cyanobacteria were more abundant in type I hypolithons, whereas Actinobacteria were relatively more abundant in types II and III hypolithons, and were the dominant group in soils. Using a probabilistic dissimilarity metric and random sampling, we demonstrate that deterministic processes are more important in shaping the structure of the bacterial community found in types II and III hypolithons. Most notably, the data presented in this study suggest that hypolithic bacterial communities establish via a successional model, with the type I hypolithons acting as the basal development state.
Similar content being viewed by others


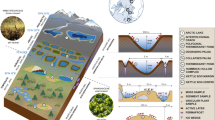
Introduction
Antarctica is regarded as one of the most ‘extreme’ environments on Earth (Cowan and Ah Tow, 2004; Convey and Stevens, 2007; Cary et al., 2010). The McMurdo Dry Valleys of Eastern Antarctica are characterized by very low levels of precipitation, episodic katabatic winds, high salt content and extremely low temperatures (Cowan and Ah Tow, 2004; Cary et al., 2010). Although it has frequently been assumed that these extreme factors would result in both low cell numbers and species diversity, recent data have supported the view that species diversity is higher than initially thought in a range of Antarctic biotopes (Cowan et al., 2002; Aislabie et al., 2006; Smith et al., 2006; Babalola et al., 2009). Evidence of potentially novel microbial species has been found in various niches ranging from permafrost to the ice-free arid terrestrial zones (Yergeau et al., 2007; Stomeo et al., 2012).
Hypolithic communities are widely distributed in hot (Schlesinger et al., 2003; Warren-Rhodes et al., 2007; Makhalanyane et al., 2013) and cold deserts (Cockell and Stokes, 2004; Smith et al., 2006; Wood et al., 2008; Pointing et al., 2009; Wong et al., 2010; Khan et al., 2011). The hypolithic ‘lifestyle’ is proposed to circumvent environmental stress as the rock provides attenuation from excessive ultraviolet, photosynthetically active radiation, freeze thaw events and enhanced water availability (Cary et al., 2010; Cowan et al., 2010b).
Bacterial community composition in hypoliths has been shown to differ from that of soil communities (Pointing et al., 2009; Khan et al., 2011; Makhalanyane et al., 2013, Stomeo et al., 2013), and to be dominated by cyanobacteria (reviewed in Chan et al. (2012) and Pointing and Belnap (2012)). Actinobacteria, Alphaproteobacteria and Gammaproteobacteria are ubiquitous in all hypoliths (Pointing et al., 2009; Wong et al., 2010), whereas Bacteroidetes, Acidomicrobia, Verrucomicrobia, Archaea, fungi or mosses represent small components (Khan et al., 2011). However, recently, two other hypolithic morphotypes have been described in the Antarctic Miers Valley (Cowan et al., 2010a): type II, dominated by fungal mycelia, and type III, dominated by bryophyte-based communities. Consequently, the cyanobacteria-dominated hypolithons are referred as type I. In an observational study, it has been suggested that the three hypolithons may represent sequential development stages (Cowan et al., 2010a). As Antarctic soils contain relatively low levels of organic carbon (Cary et al., 2010) the more obvious hypothesis is that photoautotrophic hypolithons (type I) are the primary stage in succession. The increased eutrophication of the hypolithic microenvironment resulting from cyanobacterial colonization could serve as a trigger for heterotrophic fungal colonization (type II; Cowan et al., 2010a). It was proposed that the development of the type III community may be controlled more by growth kinetics and then by nutrient status, on the basis that Antarctic mosses have very slow growth rates (Fenton, 1980) in comparison to cyanobacteria.
Hypoliths in extreme environments may be the dominant sites of primary productivity (Tracy et al., 2010), N input (Cowan et al., 2011) and the basis for the survival of whole ecosystems (Thomas, 2005). Therefore, understanding their species composition (alpha-diversity), how they vary across sites (beta-diversity) and the factors and processes that control them is of particular relevance in desert microbiology research.
Beta-diversity varies with environmental gradients (for example, productivity), environmental heterogeneity and disturbance regime, and depends on dispersal or connectivity among patches (Chase, 2003, 2007, 2010). In general, beta-diversity can be influenced by local (for example, environmental conditions and species interactions) and regional (for example, demographic stochasticity and dispersal) habitat factors (Lindström and Langenheder, 2012). Community assembly processes driven by local contemporary factors are often named niche based, species sorting or deterministic processes, whereas those driven by regional factors are so-called neutral, historical or stochastic processes. Recently, both deterministic and stochastic processes have been shown to be important in shaping the landscape distribution patterns of type I hypolithons (Caruso et al., 2011; Makhalanyane et al., 2013; Stomeo et al., 2013). In contrast, to the best of our knowledge, no study has extensively investigated the bacterial composition, factors and processes that determine the structure of types II and III hypolithic communities.
In this study, we assess bacterial beta-diversity patterns for the three different types of hypolithons to infer possible mechanisms of community assembly during succession. As biomass accumulation (productivity) is often highest in the earlier stages of succession (Fierer et al., 2010) and stochastic processes predominate in higher-productivity systems (Chase, 2010), we hypothesize that stochasticity will be stronger in cyanobacterial-relative to fungal- and moss-dominated hypolithons.
We used terminal-restriction fragment length polymorphism (T-RFLP) to show that hypoliths and soil harbor distinct bacterial communities. We then assessed bacterial community composition using 16S rRNA gene 454 pyrosequencing. As alpha-diversity varied among communities, we tested for mechanisms in community assembly by means of a probabilistic dissimilarity metric and random sampling. We present evidence that species sorting is more important in shaping the structure of types II and III relative to type I hypolithons.
Materials and methods
Sampling and soil physicochemical analysis
Samples were collected from the coastal Miers Valley (78°60′ S, 164°00′ E) region of Eastern Antarctica. The surface topology of the Miers Valley is composed of moraine of glacial and/or marine origin (Bockheim and McLeod, 2008). A total of 36 samples were collected, 9 from each of the three hypolith types and soil (that is, four habitats), and stored in sterile Whirl-Pak bags (Nasco International, Fort Atkinson, WI, USA). Equivalent amounts of hypolithic and soil samples were collected aseptically in an area of 1 km2 with similar macro-environmental conditions (that is, slope, aspect, elevation). The spatial arrangement of samples was also similar between habitats, therefore, allowing us to compare the potential influence of micro-environmental factors on beta-diversity across a similar spatial scale. Samples were maintained at −20 °C and transported to the University of the Western Cape (South Africa) and stored at −80 °C for further analysis.
The slurry technique was used to measure pH by mixing 2 g of soil with 5 ml of deionized water and allowing it to settle with a CRISON Bench meter (Crison Instruments, Barcelona, Spain). Soil chemical analysis was analyzed using a LECO Truspec Elemental Determinator at the Stellenborsch Central Analytical Facilities (Stellenbosch University, SA). Sample measurements were conducted for both anions (F−, Cl−, SO42− and NO3−) and elements (%N, %C and %S), and were analyzed using standard quality control procedures (SSSA, 1996).
DNA extraction and T-RFLP analysis
DNA was extracted from 0.5 g of sample using the MoBio PowerSoil DNA isolation kit (Mo BIO, Carlsbad, CA, USA). Adsorbed DNA was eluted in 40 μl of tris-EDTA buffer and quantified using the Nanodrop 1000 spectrophotometer (NanoDrop Products, Wilmington, DE, USA). DNA was amplified using 16S rRNA gene primers 341F-FAM and 908R (Lane et al., 1985). PCR reactions were carried out in a Bio-RAD T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA) in duplicate 50 μl reactions containing 5X Phusion HF Buffer, 10 mM dNTPs, 20 μg ml−1 bovine serum albumin, 2.5 mM MgCl2, 1 U of Phusion DNA Polymerase (Finnzymes, Espoo, Finland) and 10 ng of template DNA. Thermal cycling conditions were as follows; 3 min denaturation at 98 °C followed by 30 cycles with denaturation at 98 °C for 30 s, annealing at 55 °C for 30 s and elongation at 72 °C for 45 s with a final elongation at 72 °C for 8 min. Products were purified using the NucleoSpin Kit (Macherey-Nagel, Hoerdt, France) and digested using MspI (Fermentas, Burlington, ON, Canada). After a second purification step, electrophoretic separation of restriction fragments was conducted using an ABI3130XL (Applied Biosystems, Foster City, CA, USA). T-RFLP profiles were analyzed using Peak Scanner 1.0 (Applied Biosystems, available online https://products.appliedbiosystems.com). True peaks and fragments of comparable size were identified and binned using the software R and Perl as previously described (Abdo et al., 2006). Fragment lengths that were within 1 bp from one another were considered to represent the same operational taxonomic unit (OTU) and peaks within 3 standard deviation of the noise baseline were removed.
16S rRNA gene amplicon barcoded pyrosequencing
In order to reduce the number of samples for 454 pyrosequencing, equal amounts of DNA from each of the nine samples were pooled according to habitat (n=4). Unique four base pair multiplex identifiers were added to the primers for each sample. PCR amplification of the highly variable V3 region of the bacterial 16S rRNA gene was carried out in two steps using HotStar DNA polymerase (QIAGEN GmbH, Hilden, Germany), based on the universal bacterial primers, A8-28F (Edwards et al., 1989) and K517R (Muyzer et al., 1993). In the first PCR step, untagged primers were used in a 20-cycle reaction as described by Azmuda et al. (2012), followed by purification of the amplicons using the GenElute PCR Clean-Up Kit (Sigma-Aldrich, Copenhagen, Denmark). The second reaction was performed with 100 ng of the purified PCR amplicons as template and primers containing the 454 FLX adaptors with sample-specific multiple identifiers (Supplementary Table 1) using 10 PCR reaction cycles (Azmuda et al., 2012). The final products were purified using the Agencourt AMPure purification kit (Agencourt Bioscience Corporation (Beckman Coulter), Beverly, MA, USA) before shipment to GATC Biotech AG (Konstanz, Germany) for pyrosequencing with the GS FLX (Roche 454 Life Sciences, Branford, CT, USA) Titanium chemistry.
Pyrosequencing data were analyzed using MOTHUR (version 1.27.0; Schloss et al., 2009) following a previously established pipeline developed by Schloss et al. (2011). Briefly, the Fasta quality and flow data were extracted using the sffinfo command. In order to reduce sequences of low quality, MOTHUR uses the shhh.flows command, which is an implementation of the PyroNoise component of the AmpliconNoise suite of programs (Quince et al., 2011). The data set was simplified by obtaining the unique sequences using the unique.seqs command. An alignment was generated using the align.seqs command by aligning the data to the SILVA reference alignment ( http://www.arb-silva.de/download/arb-files/ ). The screen.seqs command was used in order to ensure that there was no overlap between the sequences. Sequences that did not match the reference alignment were removed. Identification of chimeras was through the chimera.slayer command.
Aligned sequences were used to construct a distance matrix and to group sequences into OTUs at a cutoff level of 97% in MOTHUR. The taxonomic affiliations of the OTUs were determined using the naive Bayesian rRNA classifier (Wang et al., 2007) with a confidence threshold of 80%.
Venn diagrams, rank-abundance and rarefaction curves were constructed using the R statistical package v.2.14.0 (http://www.r-project.org/). In order to compare the species diversity between the different sample types, the same number of reads per individual sample was subsampled. The Chao1 richness estimator was calculated using EstimateS (Colwell, 2009).
To estimate whether a defined bacterial community (subsidiary community) represented a subsample of another community (original community), we used a random sampling procedure (Besemer et al., 2012). OTUs were sampled from the original community with replacement until the number of OTUs in this randomly assembled community equalled the richness of the respective subsidiary community. This procedure was repeated to yield 1000 random subsamples of each original community. This procedure can be regarded as conservative as the subsidiary community was reduced to OTUs, which occurred also in the original community, therefore, increasing the chance that the subsidiary community resembles the original community.
The sequence data are available at the NCBI Sequence Read Archive under the accession number SRA058593.
Statistical analysis
T-RFLP profiles were analyzed using the vegan, gplots and labdsv packages in the software R. Bacterial richness and abiotic raw data were compared by pairwise Wilcoxon–Mann–Whitney tests after ensuring that an overall Kruskal–Wallis test was significant at P<0.05.
Abiotic data were standardized. A distance matrix was generated based on Euclidean distances, with which a non-metric multidimensional scaling ordination was performed.
In order to explore bacterial community structure, a Jaccard dissimilarity matrix was used. The structure of the bacterial community was visualized using non-metric multidimensional scaling. The effect of abiotic data in explaining variation in bacterial community structure was assessed by redundancy analysis (Legendre and Legendre, 1998). Redundancy analysis was selected as the preferred ordination, as the gradient length of the ordination axis was less than 2 standard deviation units (ter Braak and Smilauer, 2002). To examine the possible underlying mechanisms of community assembly, we used a probabilistic dissimilarity matrix (modified Raup-Crick; Chase et al., 2011). In this case, instead of representing dissimilarity among pairwise communities per se, as in most metrics of beta-diversity, the modified Raup-Crick metric expresses dissimilarity between two communities relative to the null expectation. The metric provides an indication of the degree to which deterministic processes create communities that deviate from those based on random chance alone (see Chase et al. (2011) for details). The null expectation was generated using 10 000 randomizations.
A permutational analysis of variance (Anderson, 2001) was used to test for differences in composition between habitats (‘adonis’ function in the vegan package for R), whereas permutation dispersion (Anderson, 2006) was used to test for differences in their within-habitat dissimilarity (‘betadisper’ function in the vegan package for R). The test is based on the calculation of the distances from samples to their group (habitat) centroid followed by the comparison of the average of these distances between groups, using analysis of variance. A P-value is then obtained by comparing the actual F ratio to 9999 randomly generated F ratios.
Results and discussion
In this study, hypoliths and open soil samples were assessed to investigate the bacterial community structure of hypolithic morphotypes, with the aim of demonstrating discreteness from open soil and to infer possible mechanisms of community assembly during succession.
A total of 117 T-RFLP-derived OTUs were detected of which 23 (19.6 %) were shared between the four different habitats and 54 (46.1%) were unique to the respective habitats (Figure 1a). The number of T-RFLPs per sample (α-diversity) ranged from 19 to 33. Overall, hypolithons contained higher bacterial OTU numbers than soil, and type II hypolithons appear to harbor a higher level of bacterial diversity (Table 1, Supplementary Figure 1). At a regional level (γ-diversity), 85, 65 and 57 OTUs were observed in types I, II and III hypolithons, respectively. In contrast, only 39 OTUs were observed in open soil (ca 54% reduction respective to type I hypolithons). This means that (within-habitats) compositional differences were reduced in the order type I=> type II=> type III=> open soil. Furthermore, the frequency of occurrence of OTUs differed between communities. OTU occurrences in type I hypolithons were considerably more sporadic than those in type III hypolithons and open soil (Table 1). Thus, of the OTUs observed in at least one of the type I hypolithons, each was observed in 2.5 of the nine samples, whereas of the OTUs observed in at least one of the type III hypolithons or soil samples, each was observed in 4.2 and 5.3 of the nine samples, respectively.

Venn diagram from (a) T-RFLP and (b) pyrosequencing analysis for bacterial OTUs at 97% cutoff found in hypoliths and soil samples.
Pyrosequencing analysis of partial 16S rRNA gene PCR amplicons, generated from pooled DNA (nine samples each) from each habitat type (n=4), supported findings from bacterial T-RFLP analysis. A total of 569 OTUs, cutoff defined at a 97% sequence similarity level, were identified (Figure 1b), of which 314 were singletons (Table 2). These low numbers of OTUs were not completely unexpected as lower pyrotag counts have been described in a previous study of Antarctic soils (Lee et al., 2012). We do not have a full explanation for the relatively low number of reads, in particular those from type II hypolithons, but PCR primer-related biases are well known. Thus, it is possible that the primers we used were biased against one or several groups of bacteria that were more abundant in this hypolithon type. For example, Firmicutes have been found to be especially recalcitrant to PCR amplification (for example, Ellis et al., 2003) and are commonly found in soils. The majority (156 OTUs) occurred only in type I hypolithons, whereas 77 OTUs were only in type II hypolithons and 146 OTUs only in type III hypolithons. In addition, 129 OTUs were unique to the soil and only 6 were shared by all four bacterial communities. The observation that a large number of the OTUs were unique to the habitat type could be explained in part by the fact that rarefaction curves did not reach a plateau (Supplementary Figure 2). Thus, it is possible that bacteria found in a given habitat may be present in the other habitats, albeit below the detection threshold. However, although it is likely that we did not sample the total diversity and methodological differences (for example, the PCR primers used) preclude exhaustive comparisons, these values are higher than previously found in Antarctic hypolithons (Khan et al., 2011) and Dry Valley soils (Lee et al., 2012) and suggest that bacterial biodiversity in the Miers Valley desert pavement may have been underestimated.
After the number of OTUs was resampled to the smallest effort (n=103 OTUs), richness was found to be lower in open soil samples than in hypolithons (Table 2), confirming the trend shown by the T-RFLP analysis. The rank-abundance distributions displayed a strong dominance of a few OTUs and a long tail of rare OTUs (not shown), which is a common feature among bacterial communities (Fuhrman, 2009).
A total of 13 bacterial phyla were detected of which all were represented in hypolithons and 9 were found in the open soil (Figure 2). Those phyla contributing most to the observed diversity were present in hypolithic and open soil bacterial communities; although in some cases the distribution of their relative abundance indicated a preference for one of the four habitats (Figure 2). As in previous reports, cyanobacterial phylotypes were found in both open soil and hypolithic Miers Valley samples (Wood et al., 2008) but were more abundant in type I hypolithons (Cowan et al., 2010a; Khan et al., 2011). Actinobacteria and Acidobacteria were more abundant in type III hypolithons and open soil samples, whereas Proteobacteria (mainly Alphaproteobacteria) were prevalent in types I and II hypolithons. Many members of the Proteobacteria are characterized as copiotrophic soil bacteria, that is, they compete successfully only when organic resources are abundant (Fierer et al., 2007), whereas many members of the Acidobacteria are oligotrophic. This is consistent with a model of succession in which an initial copiotrophic community, perhaps stimulated by cyanobacterial exopolysaccharides, is followed by a gradual increase in members of a more specialized oligotrophic community. In total, four (out of 6) of the most cosmopolitan OTUs were classified to the genus level. Those genera were Methylobacterium (29 OTUs), Novosphingobium (375 OTUs), Roseomonas (11 OTUs) and Sphingomonas (370 OTUs; Figure 2, Table 3). The role of these microorganisms in hypolithic communities is unknown, but Methylobacterium and Sphingomonas have been previously found in Antarctic soils (for example, Yergeau et al., 2007). Methylobacterium is resistant to both dessication and ultraviolet radiation (Romanovskaya et al., 1998) and uses C1 compounds as a facultative energy source (Green, 2006), whereas members of the genus Sphingomonas have the capacity to degrade several kinds of polymers (White et al., 1996). Such characteristics might confer an advantage to these organisms in harsh, depauperate environments. Interestingly, the most abundant OTUs in hypoliths were found in lower numbers in open soil and vice versa. This is in contrast to the composition of hypolithic communities from the Namib Desert (Makhalanyane et al., 2013) and suggests a stronger deterministic effect in shaping Antarctic hypolithons. When microenvironmental conditions (elemental chemical analysis, Supplementary Table 2) were incorporated in an non-metric multidimensional scaling ordination plot (Figure 3), hypolithic and soil bacterial communities were found to be shaped by disparate environmental factors (permutational multivariate analysis of variance (PERMANOVA): F3,35=4.08, P=0.001). However, no differences were found among the three different types of hypolithons (Table 4). A post-hoc analysis using Wilcoxon–Mann–Whitney test showed that soil samples presented lower nutrient values than hypolithons (Supplementary Figure 3), suggesting that nutritional constraints in this depauperate environment are higher for soil bacterial communities than hypolithic communities. This is entirely consistent with the different levels of primary productivity in the two habitat types (Cockell and Stokes, 2004; Cary et al., 2010), as predicted by the different photoautotrophic composition.

Line graph depicting the phylum-level distribution of bacterial OTUs (97% cutoff). The taxonomic affiliation was performed using the Ribosomal Database Project Classifier with a confidence threshold of 80%.

Non-metric multidimensional scaling ordination plot (Euclidean distance with standardized data) of microenvironmental data profiles for soil and hypolith-derived samples. Samples that are closer together are more similar in environmental conditions. The quality of the ordination is indicated by a low-stress value.
T-RFLP bacterial community analysis using Jaccard’s dissimilarity index (Figure 4) showed significant differences in community structure between hypoliths and soil samples (PERMANOVA: F3,35=6.84, P<0.001). Despite similar environmental conditions, two different hypolithic clusters were detected. The first cluster was formed by type I hypolithons, whereas a second cluster composed of types II and III hypolithons. These results were confirmed using a randomization test (Table 4), which demonstrated, surprisingly, that the bacterial composition appears to be very similar in the two very different types of eukaryotic hypolithons (types II and III). Permutation dispersion showed that type I hypolithic communities were considerably more variable in their intra-OTUs composition than the types II and III, whereas soil samples showed a very high level of compositional consistency (Table 1). Similar results were found using Bray–Curtis and Morisita–Horn dissimilarity metrics (Supplementary Figures 4 and 5). It is important to note that low abundance taxa are not detected by T-RFLP analysis. However, recent studies have shown that both 454 pyrosequencing and ‘classical’ fingerprinting methods such as Denaturing Gradient Gel Electrophoresis and T-RFLP generated comparable patterns of community composition (for example, Bengtsson et al., 2012; Besemer et al., 2012).

Non-metric multidimensional scaling ordination plot (Jaccard dissimilarity matrix) of T-RFLP profile soil and hypolith-derived samples. Points represent the composition of a community in multidimensional space, and the distance between any two points represents the difference between those two communities. Communities that are closer together are more similar in bacterial composition.
To investigate how abiotic factors affected T-RFLP patterns, we performed redundancy analysis (Figure 5). We found that sulfur, nitrate and fluoride were the most important factors explaining variability of T-RFLP patterns (P<0.001).

Redundancy analysis (RDA) biplot of bacterial diversity and microenvironmental parameters. T-RFLP analysis profiles for hypolithic and soil samples are depicted (n=36). Only the environmental variables that significantly explained variability in microbial community structure are fitted to the ordination (arrows). The direction of the arrows indicates the direction of maximum change of that variable, whereas the length of the arrow is proportional to the rate of change.
There are various possible interpretations of these results. First, it is possible that if differences in environmental parameters are higher among hypolithons, over dispersion in type I hypolithons can arise because of environmental heterogeneity. However, environmental heterogeneity, measured by permutation dispersion, showed no differences between the three types of hypolithons (Table 1). Substrate-related and/or the environmental variables that were not recorded might also influence the structure of bacterial communities. Using redundancy analysis, only 18% of the total variation in community composition could be explained by the measured variables (Figure 5), indicating that other factors may be dominant. For example, it has been shown that type I cyanobacterial hypolithons colonized rocks with approximately 50% lower photosynthetically active radiation transmittance than fungal or moss hypolithons (Cowan et al., 2010b). Salinity and porosity have been also found to be important variables determining community structure below quartz rocks (Pointing et al., 2009). Species can also alter the abiotic conditions of their environment (Jones et al., 1994). For instance, changing from a bacterial to a fungal-dominated community over the course of succession may lead to fundamental differences in nutrient availability (Cherif and Loreau, 2007). Overall, bacterial communities were strongly influenced by local environmental factors, which is consistent with the concept of habitat filtering (for example, Van der Gucht et al., 2007).
Other ecosystem features (for example, productivity) or species traits (for example, dispersal and competitive abilities) may be also important in explaining bacterial community assembly. Recent work has demonstrated that higher beta-diversity at higher productivity resulted from a stronger role for stochastic relative to deterministic assembly processes with increasing productivity (Chase, 2010). Interestingly, hypolithons dominated by cyanobacteria may be the dominant sites of primary productivity and N input (Cockell and Stokes, 2004; Tracy et al., 2010; Cowan et al., 2011). Alternatively, intensity of dispersal or complex ecological interactions can cause these patterns (Verreydt et al., 2012). However, given the small spatial scale of our study, the potential for aeolian transport and the possibility of reduced microbial competition in desert soils (Fierer et al., 2012), the patterns we observed are more likely to be affected by differences in productivity (Chase, 2010).
It is noted that most dissimilarity metrics (for example, Jaccard’s dissimilarity index) are biased by the number of species (Anderson et al., 2011). To circumvent this bias, we took advantage of a probabilistic dissimilarity metric (modified Raup-Crick; Chase et al., 2011), which measures the deviation of pairwise comparisons of community dissimilarity from the null expectation under random assembly (Figure 6). This approach allowed us to compare the beta-diversity of the different habitat types independent of differences in alpha-diversity and provides indication of the possible underlying mechanisms of community assembly (Anderson et al., 2011; Chase et al., 2011). Interestingly, this approach confirmed that the bacterial community composition was clearly different between hypolithons and open soil, and between type I and type II/type III hypolithons (PERMANOVA: P<0.001 all pairwise combinations; Table 4). Moreover, the dissimilarity values were larger among type I samples (that is, less deviant from the null expectation) relative to samples from the other three communities (Table 1, permutation dispersion), supporting the view that deterministic processes are relatively more important in explaining variations in type II/type III hypolithons and soil communities. As cyanobacteria dominate type I communities, this is consistent with conclusions by Caruso et al. (2011), who found that stochasticity is an important driver for the autotrophic component of hypolithic communities. Furthermore, simulated hypolithic communities from random sampling of the soil community demonstrate that stochastic dispersal was unlikely to shape the observed community structure of the hypolithons (Supplementary Figure 6). In contrast, we found that type II hypolithons could be originated by random sampling from type I, whereas type III could be created from type II but not from type I.

Non-metric multidimensional scaling ordination plot (modified Raup-Crick dissimilarity metric) of T-RFLP profiles for hypolith and open soil-derived samples. Communities that are closer together are more deviant from the null expectation, whereas communities that are farther apart are less deviant from the null expectation.
In summary, we have demonstrated that Antarctic hypolithons and open soils display demonstrably different bacterial community compositions. In addition, we found that beta-diversity is higher in type I hypolithons than in types II and III, suggesting that deterministic processes in the bacterial component of hypolithons might increase in the order type I=> type II=> type III. These patterns have important implications as communities of higher beta-diversity are likely to be especially vulnerable to climate change (McKnight et al., 2007). Moreover, if we assume that the effects of stochastic variation may become less important as communities develop over time (Fierer et al., 2010), these findings may validate type I hypolithons as the basal development state in the successional process (Cowan et al., 2010a). However, it is important to note that our analysis represents a snapshot in the development of these communities, with a focus on the most abundant taxa, and diversity may not have reached its maximum yet (Fierer et al., 2010). Experimental work and long-term monitoring programs of hypolithic bacterial composition involving larger sample numbers and ultra-deep sequencing may help us to resolve how the relative importance of deterministic/stochastic processes changes over time and whether or not type III are derived from type II hypolithons.
Accession codes
References
Abdo Z, Schuette UME, Bent SJ, Williams CJ, Forney LJ, Joyce P . (2006). Statistical methods for characterizing diversity of microbial communities by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Environ Microbiol 8: 929–938.
Aislabie J, Saul DJ, Foght JM . (2006). Bioremediation of hydrocarbon-contaminated polar soils. Extremophiles 10: 171–179.
Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, Freestone AL et al (2011). Navigating the multiple meanings of beta diversity: a roadmap for the practicing ecologist. Ecol Lett 14: 19–28.
Anderson MJ . (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46.
Anderson MJ . (2006). Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62: 245–253.
Azmuda N, Rahman MZ, Madsen MS, Khan SI, Birkeland N-K . (2012). Prevalence of a novel division-level bacterial lineage in lake Dhanmondi, Dhaka, Bangladesh, as revealed by deep sequencing of 16S rRNA gene amplicons. Curr Microbiol 65: 356–360.
Babalola OO, Kirby BM, Le Roes-Hill M, Cook AE, Cary SC, Burton SG et al (2009). Phylogenetic analysis of actinobacterial populations associated with Antarctic Dry Valley mineral soils. Environ Microbiol 11: 566–576.
Bengtsson MM, Sjotun K, Lanzen A, Øvreås L . (2012). Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J 6: 2188–2198.
Besemer K, Peter H, Logue JB, Langenheder S, Lindström ES, Tranvik LJ et al (2012). Unraveling assembly of stream biofilm communities. ISME J 6: 1459–1468.
Bockheim JG, McLeod M . (2008). Soil distribution in the McMurdo Dry Valleys, Antarctica. Geoderma 144: 43–49.
Caruso T, Chan Y, Lacap DC, Lau MC, McKay CP, Pointing SB . (2011). Stochastic and deterministic processes interact in the assembly of desert microbial communities on a global scale. ISME J 5: 1406–1413.
Cary SC, McDonald IR, Barrett JE, Cowan DA . (2010). On the rocks: the microbiology of Antarctic Dry Valley soils. Nat Rev Microbiol 8: 129–138.
Chan Y, Lacap DC, Lau MCY, Ha KY, Warren-Rhodes KA, Cockell CS et al (2012). Hypolithic microbial communities: Between a rock and a hard place. Environ Microbiol 14: 2272–2282.
Chase JM, Kraft NJB, Smith KG, Vellend M, Inouye BD . (2011). Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2: 24.
Chase JM . (2003). Community assembly: when should history matter? Oecologia 136: 489–498.
Chase JM . (2007). Drought mediates the importance of stochastic community assembly. Proc Natl Acad Sci USA 104: 17430–17434.
Chase JM . (2010). Stochastic community assembly causes higher biodiversity in more productive environments. Science 328: 1388–1391.
Cherif M, Loreau M . (2007). stoichiometric constraints on resource use, competitive interactions, and elemental cycling in microbial decomposers. Am Nat 169: 709–724.
Cockell CS, Stokes MD . (2004). Widespread colonization by polar hypoliths. Nature 431: 414–414.
Colwell RK . (2009). EstimateS: statistical sstimation of species richness and shared species from samples. Version 8.2. http://viceroy.eeb.uconn.edu/estimates.
Convey P, Stevens MI . (2007). Antarctic biodiversity. Science 317: 1877–1878.
Cowan DA, Ah Tow L . (2004). Endangered Antarctic environments. Annu Rev Microbiol 58: 649–690.
Cowan DA, Khan N, Pointing SB, Cary SC . (2010a). Diverse hypolithic refuge communities in the McMurdo Dry Valleys. Antarct Sci 22: 714–720.
Cowan DA, Pointing SB, Stevens MI, Craig Cary S, Stomeo F, Tuffin IM . (2010b). Distribution and abiotic influences on hypolithic microbial communities in an Antarctic Dry Valley. Pol Biol 34: 307–311.
Cowan DA, Russell NJ, Mamais A, Sheppard DM . (2002). Antarctic Dry Valley mineral soils contain unexpectedly high levels of microbial biomass. Extremophiles 6: 431–436.
Cowan DA, Sohm JA, Makhalanyane TP, Capone DG, Green TGA, Cary SC et al (2011). Hypolithic communities: important nitrogen sources in Antarctic desert soils. Environ Microbiol Rep 3: 581–586.
Edwards U, Rogall T, Blöcker H, Emde M, Böttger EC . (1989). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 17: 7843–7853.
Ellis RJ, Morgan P, Weightman AJ, Fry JC . (2003). Cultivation-dependent and -independent approaches for determining bacterial diversity in heavy-metal-contaminated soil. Appl Environ Microbiol 69: 3223–3230.
Fenton JHC . (1980). The rate of peat accumulation in Antarctic moss banks. J Ecol 68: 211–228.
Fierer N, Bradford MA, Jackson RB . (2007). Toward an ecological classification of soil bacteria. Ecology 88: 1354–1364.
Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL et al (2012). Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci USA 109: 21390–21395.
Fierer N, Nemergut D, Knight R, Craine JM . (2010). Changes through time: integrating microorganisms into the study of succession. Res Microbiol 161: 635–642.
Fuhrman JA . (2009). Microbial community structure and its functional implications. Nature 459: 193–199.
Green PN . (2006). Methylobacterium. In: Dworkin M (ed) The Prokaryotes: A Handbook on the Biology of Bacteria. Springer-Verlag: New York, pp 257–265.
Jones CG, Lawton JH, Shachak M . (1994). Organisms as ecosystem engineers. Oikos, 373–386.
Khan N, Tuffin M, Stafford W, Cary C, Lacap DC, Pointing SB et al (2011). Hypolithic microbial communities of quartz rocks from Miers Valley, McMurdo Dry Valleys, Antarctica. Pol Biol 34: 1657–1668.
Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR . (1985). Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA 82: 6955–6959.
Lee CK, Barbier BA, Bottos EM, McDonald IR, Cary SC . (2012). The Inter-Valley Soil Comparative Survey: the ecology of Dry Valley edaphic microbial communities. ISME J 6: 1046–1057.
Legendre P, Legendre L . (1998) Numerical ecology. Elsevier Science: Amsterdam.
Lindström ES, Langenheder S . (2012). Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep 4: 1–9.
Makhalanyane TP, Valverde A, Lacap DC, Pointing SB, Tuffin M, Cowan DA . (2013). Evidence of species recruitment and development of hot desert hypolithic communities. Environ Microbiol Rep 5: 219–224.
McKnight MW, White PS, McDonald RI, Lamoreux JF, Sechrest W, Ridgely RS et al (2007). Putting beta-diversity on the map: broad-scale congruence and coincidence in the extremes. PLoS Biol 5: 2424–2432.
Muyzer G, De Waal EC, Uitterlinden AG . (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59: 695–700.
Pointing SB, Belnap J . (2012). Microbial colonization and controls in dryland systems. Nat Rev Microbiol 10: 551–562.
Pointing SB, Chan Y, Lacap DC, Lau MC, Jurgens JA, Farrell RL . (2009). Highly specialized microbial diversity in hyper-arid polar desert. Proc Natl Acad Sci USA 106: 19964–19969.
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ . (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12: 38.
Romanovskaya VA, Sokolov IG, Malashenko YR, Rokitko PV . (1998). Mutability of epiphytic and soil bacteria of the genus Methylobacterium and their resistance to ultraviolet and ionizing radiation. Microbiology 67: 89–97.
Schlesinger WH, Pippen JS, Wallenstein MD, Hofmockel KS, Klepeis DM, Mahall BE . (2003). Community composition and photosynthesis by photoautotrophs under quartz pebbles, Southern Mojave Desert. Ecology 84: 3222–3231.
Schloss PD, Gevers D, Westcott SL . (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6: e27310.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Smith JJ, Tow LA, Stafford W, Cary C, Cowan DA . (2006). Bacterial diversity in three different Antarctic Cold Desert mineral soils. Microb Ecol 51: 413–421.
SSSA (1996) Methods of Soil Analysis, Part 3. Soil Science Society of America: Madison, WI.
Stomeo F, Makhalanyane TP, Valverde A, Pointing SB, Stevens MI, Cary CS et al (2012). Abiotic factors influence microbial diversity in permanently cold soil horizons of a maritime-associated Antarctic Dry Valley. FEMS Microbiol Ecol 82 (2): 326–340.
Stomeo F, Valverde A, Pointing SB, McKay CP, Warren-Rhodes KA, Tuffin MI et al (2013). Hypolithic and soil microbial community assembly along an aridity gradient in the Namib Desert. Extremophiles 17: 329–337.
ter Braak CJF, Smilauer P . (2002) CANOCO Reference Manual and CanoDraw for Windows User’s Guide: Software for Canonical Community Ordination (version 4.5). Microcomputer Power: Ithaca, NY, pp 500.
Thomas DN . (2005). Photosynthetic microbes in freezing deserts. Trends Microbiol 13: 87–88.
Tracy CR, Streten-Joyce C, Dalton R, Nussear KE, Gibb KS, Christian KA . (2010). Microclimate and limits to photosynthesis in a diverse community of hypolithic cyanobacteria in northern Australia. Environ Microbiol 12: 592–607.
Van der Gucht K, Cottenie K, Muylaert K, Vloemans N, Cousin S, Declerck S et al (2007). The power of species sorting: Local factors drive bacterial community composition over a wide range of spatial scales. Proc Natl Acad Sci USA 104: 20404–20409.
Verreydt D, De Meester L, Decaestecker E, Villena M-J, Van der Gucht K, Vannormelingen P et al (2012). Dispersal-mediated trophic interactions can generate apparent patterns of dispersal limitation in aquatic metacommunities. Ecol Lett 15: 218–226.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Warren-Rhodes KA, Rhodes KL, Boyle LN, Pointing SB, Chen Y, Liu S et al (2007). Cyanobacterial ecology across environmental gradients and spatial scales in China's hot and cold deserts. FEMS Microbiol Ecol 61: 470–482.
White DC, Sutton SD, Ringelberg DB . (1996). The genus Sphingomonas: physiology and ecology. Curr Opin in Biotech 7: 301–306.
Wong FK, Lacap DC, Lau MC, Aitchison JC, Cowan DA, Pointing SB . (2010). Hypolithic microbial community of quartz pavement in the high-altitude tundra of central Tibet. Microb Ecol 60: 730–739.
Wood SA, Rueckert A, Cowan DA, Cary SC . (2008). Sources of edaphic cyanobacterial diversity in the Dry Valleys of Eastern Antarctica. ISME J 2: 308–320.
Yergeau E, Newsham KK, Pearce DA, Kowalchuk GA . (2007). Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ Microbiol 9: 2670–2682.
Acknowledgements
We gratefully acknowledge financial support from the following organizations: the National Research Foundation (South Africa), the Research Council of Norway (the South Africa Program; grant no. 180352) and the University of the Western Cape for funding TPM, AV, N-KB and DAC. Logistical support for field research was provided by Antarctica New Zealand and the University of Waikato FRST research project (Understanding, valuing and protecting Antarctica’s unique terrestrial ecosystems: Predicting biocomplexity in Dry Valley ecosystems). We also wish to thank Dr Katharina Besemer (University of Vienna) for providing us with her R script.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Makhalanyane, T., Valverde, A., Birkeland, NK. et al. Evidence for successional development in Antarctic hypolithic bacterial communities. ISME J 7, 2080–2090 (2013). https://doi.org/10.1038/ismej.2013.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.94
Keywords
This article is cited by
-
Seasonal Variation of Hypolithic Microbiomes in the Gobi Desert
Microbial Ecology (2023)
-
Endolithic Bacterial Diversity in Lichen-Dominated Communities Is Shaped by Sun Exposure in McMurdo Dry Valleys, Antarctica
Microbial Ecology (2022)
-
Depth-Dependent Variables Shape Community Structure and Functionality in the Prince Edward Islands
Microbial Ecology (2021)
-
Hypolithic communities shape soils and organic matter reservoirs in the ice-free landscapes of East Antarctica
Scientific Reports (2020)
-
Agulhas Current properties shape microbial community diversity and potential functionality
Scientific Reports (2018)
