
Abstract
Rapid disintegration of alpine glaciers has led to the formation of new terrain consisting of mineral debris colonized by microorganisms. Despite the importance of microbial pioneers in triggering the formation of terrestrial ecosystems, their sources (endogenous versus exogenous) and identities remain elusive. We used 454-pyrosequencing to characterize the bacterial and fungal communities in endogenous glacier habitats (ice, sub-, supraglacial sediments and glacier stream leaving the glacier forefront) and in atmospheric deposition (snow, rain and aeolian dust). We compared these microbial communities with those occurring in recently deglaciated barren soils before and after snow melt (snow-covered soil and barren soil). Atmospheric bacteria and fungi were dominated by plant-epiphytic organisms and differed from endogenous glacier habitats and soils indicating that atmospheric input of microorganisms is not a major source of microbial pioneers in newly formed soils. We found, however, that bacterial communities in newly exposed soils resembled those of endogenous habitats, which suggests that bacterial pioneers originating from sub- and supraglacial sediments contributed to the colonization of newly exposed soils. Conversely, fungal communities differed between habitats suggesting a lower dispersal capability than bacteria. Yeasts putatively adapted to cold habitats characteristic of snow and supraglacial sediments were similar, despite the fact that these habitats were not spatially connected. These findings suggest that environmental filtering selects particular fungi in cold habitats. Atmospheric deposition provided important sources of dissolved organic C, nitrate and ammonium. Overall, microbial colonizers triggering soil development in alpine environments mainly originate from endogenous glacier habitats, whereas atmospheric deposition contributes to the establishment of microbial communities by providing sources of C and N.
Similar content being viewed by others


Introduction
Knowledge about the dispersal of microorganisms and how they colonize newly exposed habitats is essential for understanding the biogeographical patterns of microbial diversity (Martiny et al., 2006; Tedersoo et al., 2012). Most alpine glaciers are currently retreating leading to formation of new terrestrial ecosystems. Glacier ecosystems have been used extensively to investigate dynamics of soil formation in relation to shifts in microbial communities (for example, Bernasconi et al., 2011; Zumsteg et al., 2012; Bajerski and Wagner, 2013; Rime et al., 2015). They also represent ideal ecosystems to study natural establishment of microbial communities in terrestrial environments (Edwards et al., 2014). When glacier ice melts, entrapped rocks, sediments and fine debris are released and microbial pioneers present on these debris initiate soil formation by weathering rock minerals and incorporating organic matter (Frey et al., 2010; Smittenberg et al., 2012).
Despite the importance of pioneer microorganisms in triggering soil formation, their origin and identity are largely unknown (Bradley et al., 2014). Mineral debris released after glacier retreat is exposed to continuous atmospheric input (exogenous source) of microbes (Womack et al., 2010) through precipitations (Zhang et al., 2010; Šantl-Temkiv et al., 2012) and dry deposition, such as wind-blown dust and plant debris (Chuvochina et al., 2011; Bowers et al., 2012; DeLeon-Rodriguez et al., 2013). The atmosphere in particular is often wrongly considered sterile because of harsh ultraviolet (UV) radiation and extreme C and nutrient limitations (Womack et al., 2010). However, recent studies have reported that the atmosphere harbours complex and viable microbial communities (Vaïtilingom et al., 2010; Šantl-Temkiv et al., 2012; Stres et al., 2013). For example, Amato et al. (2005) isolated active bacteria and fungi from cloud droplets. Similarly, Šantl-Temkiv et al. (2013) reported that viable methane-oxidizing bacteria were found in rain and Favet et al. (2013) retrieved living algae (for example, Chlorella) and fungi (for example, Aspergillus) in wind-blown desert dust. These studies thus suggest that microorganisms may be transported through the atmosphere and may represent a non-negligible reservoir of microbes able to colonize recently deglaciated mineral debris. In contrast to these exogenous sources, microorganisms already present at the surface of the glacier, entrapped in ice, transported by glacial water runoff and thriving under basal ice (endogenous sources) can also potentially colonize deglaciated debris and participate in soil formation (Edwards et al., 2014). Hamilton et al. (2013) reported that viable microorganisms thrive under ice. In addition, several studies showed that cryoconite holes and supraglacial debris are hot-spots of microbial diversity on top of glaciers (Anesio et al., 2009, 2010; Edwards et al., 2013b; Franzetti et al., 2013) and may thus constitute part of the microbial pioneer communities in recently deglaciated barren soils.
Although these exogenous and endogenous sources have been investigated separately, their relative contribution to microbial colonization of mineral debris recently exposed after glacier retreat has not been assessed yet. To address this gap, we simultaneously characterized the microbial communities in exogenous and endogenous sources and compared them with the microbial communities found in barren soils formed after the retreat of the Damma glacier (Switzerland). In particular, we used barcoded 454-pyrosequencing of archaeal, bacterial and fungal ribosomal markers. Our study specifically addresses the following questions: (1) What is the origin (exogenous versus endogenous sources) of the microbes colonizing newly formed terrain after glacier retreat? (2) Which microbial taxa are characteristic of the exogenous and endogenous sources? (3) Which environmental factors shape the microbial taxa characteristic of these habitats?
Materials and methods
Site description
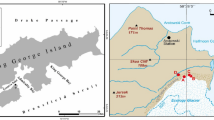
Samples were collected in the Damma glacier catchment in the Central Alps of Switzerland. This site was selected because it has been studied extensively in various scientific disciplines, including hydrology, botany, soil carbon dynamics and microbiology (Bernasconi et al., 2011; Menon et al., 2014), and thus detailed information about the ecosystem dynamics in this glacier forefield was available. The climate in the catchment area is characterized by annual precipitation of 2200 mm and high seasonal temperature fluctuations, ranging from −8 °C to 4 °C, with an annual average temperature of 2 °C (Bernasconi et al., 2011). The growing season lasts from June to September. Before the glacier terminus, the initial soils sampled in our study are located 20 m away from the glacier tongue (deglaciated for approximately 2 years) and consist of barren sandy rocks with low carbon and nitrogen contents and are mainly characterized by lichens, algae and mosses (Bernasconi et al., 2011).
Experimental procedures
In our sampling approach, snow (referred hereafter to as Snow), as well as dry and wet atmospheric summer deposition (Deposition) were considered exogenous sources of microbial colonizers, whereas basal glacier ice (Ice), glacier stream water (Glacier stream), sub- (Subglacial sediment) and supraglacial (Supraglacial sediment) sediments were considered endogenous sources of microbial colonizers. Microbial communities living in snow may originate from more distant soils, aerosolized and deposited with snow (Cameron et al., 2015). We therefore considered this habitat as an exogenous source of microbial pioneers. Owing to the fact that the Damma glacier ice is covered by sediments, we could not identify cryoconite holes. We therefore sampled mineral debris and referred to them as supraglacial sediments. Snow was sampled on 23 June 2014. Barren soils, considered as a potentially colonizable habitat, were sampled before snow melt (Snow-covered soil; collected on 23 June 2014) and 3 weeks after snow melt (Barren soil; collected on 10 July 2014) to determine whether microbial pioneers present in snow were also found in barren soils after snow melt. We sampled all other habitats 3 weeks (collected on 10 July 2014) after snow melt, as recommended by Musilova et al. (2015). Detailed information about the sampling procedure and sample locations is given in Supplementary Table S1 and Supplementary Figure S1.
Briefly, approximately 2 litre snow surfaces (0–2 cm depth) were sampled in triplicate during the melting phase, as described by Lazzaro et al. (2015), with EtOH-cleaned laboratory spoons in autoclaved polyethylene (PE) bags. After sampling of the snow patches, we removed snow aseptically to collect around 100 g of barren soil (0–2 cm depth). After snow melt, around 100 g of barren soil (0–2 cm depth) were sampled at the same sites, which were marked with sticks during snow sampling. We installed three sets of sampler pair to collect atmospheric deposition at the same locations as the snow and soil samples. Each sampler consisted of a sterile (acid and EtOH rinsed) 1 litre PE bottle coupled with a sterile 12- cm diameter PE funnel fixed on a stake 1 m above the ground. The samplers were left open for 2 weeks (from 30 June 2014 to 9 July 2014) to collect passively atmospheric dust at ambient temperature and were sampled directly after a rain event (on 9 July 2014) in order to recover both atmospheric dry and wet inputs while limiting aftergrowth. Around 1 litre of rain per bottle resulting in approximately 2 litre of rain per sampler pair were collected in July after a Saharan dust event, and in August (left open from 31 July 2014 to 9 August 2014; collected on 9 August 2014), when no Saharan dust event was detected at the Jungfraujoch research station (Collaud Coen et al., 2003). Owing to the absence of significant differences in α- and β-diversities and physico-chemical factors between the two sampling times (data not shown), we averaged these samples and treated them as pools to represent atmospheric deposition throughout the growing season. We collected three replicates of 2 litre of stream water at the mouth of the glacier terminus in individual sterile 1 litre PYREX bottles previously rinsed three times with Milli-Q water before being autoclaved. We accessed basal glacier ice by penetrating into a cave naturally formed by glacier melting. Snow blocked access to the cave until the end of June (beginning of snow melt), ensuring us that the ice had not been exposed to atmospheric contaminants. Twelve subsamples of approximately 2 litre of ice were collected aseptically with an ice pick in autoclaved PE bags, following methods used in previous studies (Felip and Sattler, 1995; Hamilton et al., 2013). In the same cave, 100 g of subglacial sediments deposited during ice melt were sampled aseptically in triplicate. Approximately 200 g of supraglacial sediments were collected in triplicate on the glacier ice above the cave. All samples were kept cold during transportation, stored and/or melted at 4 °C overnight. Melted samples of snow and ice, glacier stream water and atmospheric deposition were each divided into two portions and filtered the day after sampling. The first portion was filtered through 0.22 μm pore-size water filters (MoBio, Carlsbad, CA, USA) for DNA extraction, whereas the other portion was filtered through 0.44 μm pore-size water filters (MoBio) for chemical analyses. Filters (for DNA extraction) and filtrates (for chemical analyses) were kept frozen at −20 °C. Soil and sediment samples were homogenized through a 2 mm sieve and divided into two subsamples. The first subsamples were weighed and frozen at −20 °C in DNA extraction buffer (MoBio), whereas the second subsamples were oven-dried overnight at 105 °C and extracted with Milli-Q water (1:5 m/v ratio) by using an overhead shaker overnight. Soil and sediment slurries were first filtered through glass fibre filters (Whatman GF/F glass microfiber filter, GE Healthcare, Little Chalfont, UK) precombusted at 450 °C. These subsamples were then filtered through 0.44 μm pore-size filters to obtain a particle size similar to that of the deposition, snow, ice and glacier stream filtrates. All filtrates were collected in acid-cleaned 100 ml PE bottles and pH was determined with a pH-metre (FEP20-FiveEasy Plus, Mettler Toledo GmbH, Switzerland). Nitrate (NO3−), phosphate (PO43−), sulphate (SO42−) and chloride (Cl-) concentrations were measured by ion chromatography with a IC:DX-120 chromatograph (Dionex Corp., Sunnyvale, CA, USA), and ammonium (NH4+) concentrations was measured photometrically with a FIAS 300 (PerkinElmer, Waltham, MA, USA) and dissolved organic carbon (DOC) and nitrogen (DON) concentrations were measured with a TOC-V analyser (Shimadzu, Tokyo, Japan).
DNA extraction, PCR reactions and 454-pyrosequencing
Total DNA from soils and sediments (approximately 1 g) was extracted with the UltraClean Soil DNA extraction kit (MoBio), whereas the 0.22 μm pore-size filters were extracted with the RapidWater DNA Isolation Kit (MoBio) according to the manufacturer protocols. DNA was quantified with PicoGreen (Invitrogen, Carlsbad, CA, USA) and stored at −20 °C.
As the ice samples yielded small amounts of DNA, four individually extracted subsamples were pooled, precipitated and resuspended in 50 μl elution solution according to the manufacturer instructions to form three independent replicates. Amplicons were generated using methods similar to those applied by Hartmann et al. (2014) and Rime et al. (2015). Briefly, DNA was pre-treated with 1 μg bovine serum albumin ml−1 at 95 °C for 5 min before PCR reactions to bind PCR inhibitors. The regions V1–V3 and V3–V5 of the bacterial and archaeal small-subunit ribosomal RNA genes, respectively, as well as the ITS-2 region of the fungal ribosomal operon were amplified by performing three PCRs with 10 ng template DNA and the HotStar Taq amplification kit (Qiagen, Hilden, Germany) in a final volume of 25 μl. All PCRs were conducted on a standardized amount of DNA to account for different microbial cell numbers in the habitats sampled. PCR conditions are given in the Supplementary Information. The purified PCR products with different barcoded primers were pooled in equimolar concentrations and shipped to the sequencing facility (Genome Quebec Innovation Center, Montreal, Canada) where they were unidirectionally sequenced with the 454-pyrosequencing GS-FLX Titanium technology (Roche 454 Life Sciences, Branford, CT, USA) from the primers 27f, 349f and ITS4.
Sequence analyses
Bioinformatics quality checks of the pyrotagged reads were performed in MOTHUR v.1.32 (Schloss et al., 2009) according to the established procedure recently described (Hartmann et al., 2014; Rime et al., 2015). A detailed description of the sequence curation procedure is given as Supplementary_Materials and Methods.doc. Curated sequences as well as operational taxonomic unit (OTU) tables are available in the Supplementary Information. Less than 6% of archaeal sequences could be classified at the phylum level (see Discussion section for details). Therefore, our in-depth analyses only focused on the bacterial and fungal sequences.
Quantitative real-time PCR reactions
Relative abundances of bacterial 16S and fungal ITS rRNA gene copies were determined by quantitative real-time PCR on an ABI7500 Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) as described in Rime et al. (2015). Quantitative real-time PCR reactions were performed using 2 ng DNA in a total volume of 25 μl containing 0.5 μM of each primer, 0.2 mg ml−1 bovine serum albumin and 12.5 μl of QuantiTect SYBR Green PCR master mix (Qiagen). Cycling conditions were similar to those used for the pyrosequencing approach. Standard curves (correlations ⩾0.997) were obtained using 10-fold serial dilutions (10−1 to 10−9 copies) of plasmids generated from cloned targets (Frey et al., 2011).
Statistical analyses
We estimated the observed richness (Sobs), Shannon diversity (H) and Gini coefficient (G), a measure of inequality where values close to 0 correspond to even communities (Dorfman, 1979; Edwards et al., 2013b), based on evenly rarefied OTU matrices (Bacteria: 3764 sequences; Fungi: 675 sequences). We assessed differences in α-diversity indices between exogenous and endogenous sources as well as among different soil habitats by performing a one-way analysis of variance (ANOVA) in R v.2.15 (R development Core Team, 2012). Tukey's Honestly Significant Difference (Tukey HSD) post hoc tests were conducted to examine pairwise differences among habitats with the HSD.test function implemented in the agricolae package in R (De Mendiburu, 2012). As the library sizes differed among samples, we also investigated rarefaction curves. Rarefaction curves for bacterial and fungal observed richness were generated by pooling all sequences for each habitat with the rarefaction.single function as implemented in MOTHUR using 1000-fold resampling without replacement. Bray–Curtis dissimilarities were calculated based on square-root transformed relative abundances of OTUs. Differences in community structures (β-diversity) were assessed by conducting a permutational ANOVA (PERMANOVA, number of permutations=99 999) with the function adonis and an analysis of similarity (ANOSIM, number of permutations=99 999) implemented in the vegan R package (Oksanen et al., 2012) and displayed with principal coordinate analysis ordinations. As significant differences detected by PERMANOVA may arise because of within-habitat variation (Hartmann et al., 2012), we performed an analysis of homogeneity of group dispersion (Dispersion) (Legendre and Anderson, 1999) by using the function betadisper implemented in the vegan package (Oksanen et al., 2012). In our multivariate analyses, rare OTUs, that is, OTUs represented by one (singletons) or two (doubletons) sequences, were not removed, unless mentioned otherwise, because they may represent real organisms and they do not substantially influence changes in β-diversity (Gobet et al., 2010). Changes in α- and β-diversities of the most abundant (relative abundance >1% of the total sequence number) phyla or classes among the Proteobacteria were assessed using analyses similar to that applied to the total communities. Differences in relative abundances of the most abundant taxa were determined by performing a one-way ANOVA followed by Tukey HSD tests. All graphs were generated in R with the graphics (R development Core Team, 2012) or ggplot2 (Wickham, 2009) packages.
We additionally conducted an indicator species analysis to identify taxon–habitat association patterns. This procedure identifies OTUs as indicator species independently from their abundance in the total data set. However, for this analysis, single- and doubleton OTUs were removed as they hold little indicator information. This analysis was done with the multipatt function (number of permutations=99 999) implemented in the indicspecies R package (De Cáceres et al., 2010). To account for multiple testing, P-values were corrected by calculating false discovery rates (q-values) with the q-value function implemented in the BiocLite R package (Dabney and Storney, 2014). Indicator OTUs with q<0.05 were considered significant. Indicator taxa were represented in bipartite networks by using the edge-weighted spring embedded algorithm layout implemented in Cytoscape v.3.0.2 (Shannon et al., 2003) where point biserial correlation values, a measure of species–habitat association, were used to weight the edges between nodes constituting the habitats and indicator OTUs (Hartmann et al., 2015). We further mapped these indicator OTUs on taxonomic networks generated in Cytoscape v.3.0.2 to investigate potential taxa–habitat associations (Hartmann et al., 2015). Indicator OTUs classified at the genus level were displayed in a taxonomic tree generated in iTOL (Letunic and Bork, 2011) together with the positive point biserial correlation values associated with each habitat. To find patterns of co-occurrence among OTUs characteristic of the habitats studied, we analysed correlations among the relative abundances of all indicator OTUs (co-correlations) by calculating Spearman rank correlation values (Spearman, 1904) in R with the function corr.test implemented in the package psych. Multiple testing was accounted for by correcting P-values with a false discovery rates of 5% (q-value<0.05). Bacterial and fungal indicator OTUs that were significantly co-correlated were displayed in networks with the edge-weighted spring embedded algorithm layout implemented in Cytoscape where Spearman correlation values were used to weight the edges between the nodes representing the OTUs (Hartmann et al., 2015).
Differences in relative abundances of ribosomal gene copies and in environmental factors between habitats were assessed by conducting a one-way ANOVA followed by Tukey HSD tests. To investigate the effect of environmental factors on microbial communities, we conducted a DistLM analysis (Legendre and Anderson, 1999) using a step-wise selection procedure with the adjusted-R2 criterion selection in PRIMER6+ (Clarke and Gorley, 2006). Selected factors were used to build distance-based redundancy ordinations (db-RDA). Similarly to the indicator species analysis, we calculated Spearman rank correlation values between environmental factors used in db-RDAs and the relative abundances of all OTUs with the corr.test function implemented in the psych R package (Revelle, 2014). This approach enabled us to identify significant associations between particular OTUs and environmental factors. We accounted for multiple testing by correcting P-values using a false discovery rate of 5% (q<0.05). OTUs classified at the genus level were displayed in a taxonomic tree generated with iTOL where the positive Spearman rank correlation values between OTUs and the selected environmental factors were mapped.
Results
Environmental factors and relative abundances of ribosomal gene copies
All environmental factors except for NH4+ and PO43− concentrations varied between habitats (Table 1). DON, NO3− and SO42− concentrations were highest in deposition and DOC was highest in barren soil, and pH was highest in deposition and glacier stream. Relative abundances of bacterial 16S and fungal ITS gene copy numbers significantly differed between habitats (Table 2) and were highest in barren soil, snow-covered soil and supraglacial sediments and were lowest in ice (Figure 1).

Changes in bacterial and fungal α-diversity indices among the habitats investigated. Horizontal lines represent the median, whereas the boxes represent the interquartile range of the first and third quartiles. The vertical lines (whiskers) represent the maximal and minimal values. Points within each boxplot represent the means (n=3). Letters indicate differences between individual means assessed with Tukey HSD post hoc tests.
Community compositions
We obtained 174 096 (7254±1957 per sample) bacterial 16SV1-V2 and 157 292 (5826±2501 per sample) fungal ITS high-quality sequences resulting in 3942 (624±346 per sample) bacterial and 1611 (157±66 per sample) fungal OTUs. The bacterial communities were dominated by Proteobacteria (56%) followed by Bacteroidetes (13%), Cyanobacteria (8%), Actinobacteria (7%), Acidobacteria (4%), Chloroflexi (3%), Gemmatimonadetes (2%), Planctomycetes (1%) and TM7 (2%), whereas <1% were unclassified. Combined, the remaining 35 phyla represented 3.5% of the total bacterial sequences. Among Proteobacteria, Betaproteobacteria was the most abundant class (32%) followed by Alphaproteobacteria (12%), Gammaproteobacteria (9%) and Deltaproteobacteria (2%). The fungal communities consisted of Ascomycota (64%), Basidiomycota (23%) and unclassified fungi (12%), whereas Chytridiomycota, Glomeromycota and Zygomycota represented <1% of the total fungal sequences.
Alpha-diversity
For bacteria and fungi, as well as their most abundant taxa, α-diversity indices differed between endo- and exogenous sources of microbial colonizers and among soil habitats (Table 2). Observed richness (Sobs) and Shannon diversity (H) of bacteria were highest in barren soil and lowest in deposition and snow (Figure 1). The bacterial Gini coefficient (G) was lowest in barren soil and highest in deposition and snow indicating that the bacterial communities in deposition and snow were dominated by a few abundant OTUs. Fungal Sobs, H and G differed between subglacial sediments, barren soils and snow-covered soils. Among the most abundant bacterial and fungal phyla or classes of Proteobacteria, Sobs and H were generally highest in barren soil and snow-covered soil, whereas G was lowest in barren soil and highest in deposition and snow (Supplementary Figure S2). Rarefaction curves showed that the α-diversity differed among habitats (Supplementary Figure S3). The bacterial α-diversity was highest in snow-covered and barren soils and lowest in snow and deposition. Similarly, the fungal α-diversity in deposition was lowest. In contrast to bacteria, fungal α-diversity was highest in supraglacial sediments and similar in snow-covered soils and snow.
Beta-diversity
Principal coordinate analysis ordinations and PERMANOVA revealed that the structures of the total bacterial and fungal communities as well as their most abundant taxa also changed among the different habitats investigated (Figures 2a and b,Table 2 and Supplementary Figure S4). These results were supported by the ANOSIM statistics. However, the analysis of within-habitat community dispersion showed that structural differences in Chloroflexi, Cyanobacteria, Planctomycetes, Alphaproteobacteria, Deltaproteobacteria, TM7 as well as the total fungi, Ascomycota and Basidiomycota were at least partially caused by different within-habitat heterogeneities. Further, the relative abundances of all taxa investigated significantly varied between habitats. For example, the relative abundance of Acidobacteria and Deltaproteobacteria was highest in barren soil, whereas that of Cyanobacteria and Basidiomycota was highest in snow (Supplementary Figure S5).

Differences in total bacterial (a) and fungal (b) community structures shown by principal coordinate analysis ordinations (PCOs). The variance explained by each axis is given in parentheses. Colours and symbols are described in the figure. Bacterial (c) and fungal (d) bipartite association networks showing significant (q<0.05) positive associations between indicator OTUs and specific habitats, represented as nodes. Edges, that is, the connections between the habitat and an OTU, were weighted according to the point biserial correlation values. Node sizes represent the square-root of the relative abundances of each OTUs. Colours and symbols used to illustrate association between habitats and OTUs are identical to those in the PCOs. In addition, white hexagons represent OTUs that are significantly associated with more than one habitat (shared OTUs). The number of indicator OTUs is mentioned for each habitat.
We identified 670 bacterial and 176 fungal indicator OTUs, which were significantly (q<0.05) associated with a particular habitat or a combination of habitats (Supplementary Table S2) and were represented in bipartite networks (Figures 2c and d). The structure of the bipartite networks resembled the principal coordinate analysis ordinations, showing strong differences in indicator taxa characteristic of exogenous sources (deposition in particular) endogenous sources and soil habitats. Four hundred bacterial indicator OTUs were shared between the different habitats, whereas 48 fungal indicator OTUs were shared between habitats. Among the bacterial indicator, OTUs shared among habitats, 94 OTUs (14% of the total number of indicator OTUs) were shared between newly exposed soils (barren and snow-covered) and subglacial sediments, 71 OTUs (11%) between newly exposed soils and supraglacial sediments, 55 OTUs (9%) between newly exposed soils and glacier stream, and 20 OTUs (3%) between newly exposed soils and ice. The evaluation of co-correlation patterns of indicator OTUs revealed that bacterial indicator OTUs characteristic of exogenous sources (deposition or snow) were negatively correlated with indicator OTUs of other habitats (soils or endogenous sources), whereas fungal indicator OTUs associated with snow and supraglacial sediments were positively co-correlated (Figure 3,Supplementary Table S3).

Networks of significantly (q<0.05) co-correlated bacterial and fungal indicator OTUs. The nodes represent OTUs while the edges connecting nodes are positive Spearman rank correlation values. Node sizes represent the square-root of the relative abundance of each OTU and node shapes illustrate the habitat type with which each OTU is associated. White hexagons represent OTUs that are associated with more than one habitat (shared OTUs).
Among the indicator OTUs, we identified 139 bacterial and 47 fungal indicator OTUs that were classified at least down to the genus level (Figure 4), representing 42% and 47% of the total bacterial and fungal sequences, respectively. Most of the OTUs affiliated to genera belonging to Bacteroidetes were positively associated with glacier stream while no other particular phylum–habitat association could be observed (Supplementary Figure S6). For example, representatives of genera positively associated with deposition were distributed among Actinobacteria (Rhodococcus and Mycetocola), Bacteroidetes (Sediminibacterium), and Alpha- (Methylibium and Sphingomonas), Beta- (Variovorax) and Gammaproteobacteria (Pseudomonas). Similarly, indicator OTUs associated to barren soil were affiliated to various phyla, such as Acidobacteria (Candidatus Koribacter), Firmicutes (Desulfosporosinus), Alphaproteobacteria (Telmatospirillum), Gammaproteobacteria (Rhodoferax) and Verrucomicrobia (Opitutus). However, at the genus level, some patterns emerged. For example, representative OTUs affiliated to Geobacter (Proteobacteria) were only associated with barren soil and snow-covered soil. OTUs belonging to Candidatus Koribacter (Acidobacteria) and Desulfosporosinus (Firmicutes) were associated with barren soil. Similarly for the fungi, representatives belonging to Leucosporidium and Rhodotorula (both Basidiomycota) were specifically prominent in snow and supraglacial sediments, whereas OTUs affiliated to Aureobasidium (Ascomycota) and Mucor (Zygomycota) were associated with deposition.

Taxonomic tree representing the bacterial and fungal indicator OTUs classified at the genus level. The phyla within each domain are mapped on the branches of the taxonomic tree as numbers. Circles mapped in the outer rim of the tree represent the positive point biserial correlation values between each OTU and the habitats investigated.
The environmental factors that explained most of the variability in bacterial and fungal β-diversities were DOC, DON, Cl-, pH and SO42- concentrations (Figures 5a and b, Table 3) and accounted for 42% and 51% of the overall variability observed in the bacterial and fungal communities, respectively. We identified 137 bacterial and 37 fungal OTUs that were significantly correlated (q<0.05) with the selected environmental factors (Supplementary Table S4). Thirty-nine of the bacterial and 12 of the fungal OTUs significantly associated with these environmental factors were classified at the genus level (Figure 5c). OTUs affiliated to Acidobacteria, Cyanobacteria, OP10 and Planctomycetes were positively correlated with DOC and Cl-, whereas OTUs affiliated to Actinobacteria, Bacteroidetes (except for an OTU related to the genus Segetibacter) and Ascomycota (except for OTUs related to Cladiophialophora and Trichoderma) were positively correlated with SO42−, pH and DON. Among Alphaproteobacteria, OTUs affiliated to the genera Hyphomicrobium and Rhodoplanes were positively correlated with DOC and Cl-, whereas the other OTUs were correlated with SO42−, pH and DON.

Distance-based redundancy ordinations (db-RDA) built based on selected environmental variables that constrained most of the variability of the bacterial (a) and fungal (b) community structures. OTUs classified at the genus level that were positively correlated with at least one of these environmental factors were represented in a taxonomic tree (c) where the circles in the outer rim represent the Spearman rank correlation values between the OTUs and the environmental factors.
Discussion
This study investigated the microbial communities present in various habitats of the Damma glacier catchment (endogenous sources: ice, glacier stream, sub- and supraglacial sediments; exogenous sources: aeolian dust, rain and snow). We used 454-pyrosequencing to identify microorganisms that may colonize mineral debris present at the surface of recently deglaciated soils. We found that α-diversity in the investigated habitats was in accordance to values reported by others in glacial ecosystems (in high-elevation streams: Wilhelm et al., 2013; in glacier debris: Franzetti et al., 2013; in barren soils: Brown and Jumpponen, 2014; Rime et al., 2015). Despite the occurrence of harsh conditions in the investigated habitats, such as high UV radiation, daily and seasonal temperature fluctuations, as well as paucity of nutrients and C, our study supports the existing body of evidence that alpine environments have a high genetic pool of microorganisms. Despite all precautions applied during sequence processing and statistical analyses, conclusions concerning microbial α-diversity and, to a lesser extent, β-diversity should be considered carefully given the difficulty to properly explore microbial diversity (Hughes et al., 2001). Taxon–habitat association patterns revealed distinct dispersal patterns between bacterial and fungal communities (Figures 2 and 3). Communities from endogenous sources shared many bacterial indicator OTUs with soils but only a few with snow and atmospheric deposition. Conversely, fungal communities were generally distinct, with a few indicator OTUs shared among habitats. Interestingly, the fungal community in snow resembled that of supraglacial sediments although these habitats were not in direct contact during sampling (Supplementary Table S1). Advected sediment-associated fungi may have been deposited on the snow surface, which would explain these similarities. Our findings suggest that atmospheric deposition of microorganisms is not the dominant sources of microbial colonizers of recently deglaciated mineral debris for the period sampled. However, inputs of C, N (as DON, nitrate and ammonium) and other nutrients (for example, sulphate and phosphate) from atmospheric deposition represent important inputs to nutrient and C pools, which are known to be small in barren soils (Bernasconi et al., 2011; Smittenberg et al., 2012). Therefore, these inputs may facilitate the establishment of pioneer microbial communities in the mineral debris (Frey et al., 2010, 2013; Brunner et al., 2011).
The simultaneous investigation of bacterial and fungal communities present in atmospheric deposition, endogenous glacial habitats and barren soils revealed interesting patterns. Sixty percent (400) of the bacterial indicator OTUs were shared between at least two different habitats, whereas only 27% (48) of the fungal indicator OTUs characterized more than one habitat, suggesting different dispersal patterns between bacterial and fungal communities. Most of the bacterial OTUs classified at the genus level that were characteristic of the barren soils were also present in endogenous habitats or snow-covered soils. This finding is in agreement with previous studies that reported similar bacterial communities in different but spatially connected habitats (Hodson et al., 2008; Řeháková et al., 2010; Edwards et al., 2013a). At a larger biogeographical scale, bacteria affiliated to the genus Polaromonas have been recorded throughout the cryosphere worldwide (Priscu and Christner, 2004; Darcy et al., 2011; Larose et al., 2013; Hell et al., 2013). In contrast, 73% (154) of the fungal indicator OTUs in our study were only associated with one habitat, indicating a lower dispersal efficiency than bacteria. Similar to our results, Naff et al. (2013) observed that fungal zoospores affiliated to Chytrids differed between geographically close snowpacks. Further, Edwards et al. (2013b) reported that fungal community structures differed between moraine debris, tundra soils and cryoconite holes in three different Arctic glaciers. Their study also identified unknown fungi in cryoconite holes that were not retrieved from surrounding moraine debris or tundra soils, indicating that cryoconite holes may represent specific reservoirs of fungal biodiversity. We suggest that the difference in dispersal patterns between bacterial and fungal communities may be due to differences in the size and physiological capabilities of bacteria and fungi as discussed by Schmidt et al. (2014). Dispersal is thought to be a key factor driving biogeographical patterns of bacterial communities (Lindström and Langenheder, 2012). Overall, our study indicates that differences in dispersal ability between bacterial and fungal communities may be a crucial factor influencing microbial connectivity among habitiats and, in a broader context, biogeographical patterns of microorganisms.
One aim of this study was to identify whether potential microbial colonizers of mineral debris were deposited from the atmosphere. Microorganisms travelling through the atmosphere represent perfect immigrant candidates (Pearce et al., 2009) to establish in nutrient-poor soils formed after glacier retreat because they can thrive and remain active under high UV exposure, osmotic stress and C depletion (Amato et al., 2007; Šantl-Temkiv et al., 2012; Šantl-Temkiv et al., 2013). We found, however, that atmospheric bacterial and fungal communities deposited in the Damma glacier catchment were different from those of soils. Atmospheric bacteria and fungi can originate from various sources, such as soils, water and plants (Bowers et al., 2009, 2012), and can be transported through the atmosphere attached to dust particles (Favet et al., 2013) or freely as single cells participating in ice nucleation processes (Bowers et al., 2009; Šantl-Temkiv et al., 2012). Recently, Hervàs et al. (2009) and Barberán et al. (2014) investigated bacterial communities in Saharan dust deposited in an oligotrophic Pyrenean lake and found that few microbial immigrants could survive after being deposited. By identifying indicator OTUs and analysing co-correlation patterns among them, we could highlight OTUs causing changes in multivariate patterns (Hartmann et al., 2012, 2014, 2015; Rime et al., 2015). Co-correlation patterns among bacterial and fungal indicator OTUs revealed that indicator OTUs characteristic of atmospheric deposition did not share the same habitats as the other indicator OTUs. In addition, bacterial and fungal OTUs characterizing atmospheric deposition (rain and aeolian dust) consisted of either pathogenic epiphytic (Aureobasidium) or highly versatile microorganisms (Methylobacterium, Pseudomonas and Sphingomonas) predominantly associated with plants (Vorholt, 2012). Representatives of these bacterial genera have also been found in cloud droplets (Amato et al., 2005), in hailstones (Šantl-Temkiv et al., 2012) and in aerosols collected at high altitude (Bowers et al., 2012). Similarly, OTUs present in snow, such as representatives of the Chlamydomonadaceae and Acetobacteraceae families, were mostly absent from other endogenous glacial habitats or barren soils. Despite their potential ability to survive and to be transported through the atmosphere, these microorganisms may be outcompeted when deposited on recently deglaciated barren soils. The successful colonization and establishment in a new environment by external microbial communities depend on the order of arrival of the colonizers, also referred to as the 'priority effect'. The priority effect has already been used to explain successful colonization of new habitats by microorganisms in aquatic ecosystems (Symons and Arnott, 2014; Incagnone et al., 2014; Naselli-Flores et al., 2016). Further, in microcosm experiments, Werner and Kiers (2015) showed that the arrival order of arbuscular mycorrhizas influenced the successful colonization and future development of root-associated fungal communities in soil. Although we investigated a different colonizable ecosystem (glacier), our results are in agreement with these studies. This suggests that ecological niches of aerosolized microbes may already be occupied by glacier-related microorganisms when they are deposited, hindering thus successful establishment. Overall, our study indicates that the priority effect has a role in determining initial ecological trajectories of microbial communities during soil formation.
We found that many bacterial indicator OTUs were associated with soils and endogenous glacial habitats (ice, glacier stream, sub- and supraglacial sediments). For example, OTUs affiliated to the genera Bradyrhizobium, Hyphomicrobium, Methylibium and Rhodoplanes were characteristic of sub- and supraglacial sediments, barren soils and snow-covered soils. Members of these genera are commonly isolated from various habitats (soils, wastewater-treatment plants and aquatic environments). They have extremely diverse metabolisms, ranging from facultative methylotrophy, for example, for Methylibium and Hyphomicrobium, (Holm et al., 1996; Nakatsu et al., 2006; Stackebrandt et al., 2009) to facultative photoorganotrophy, for example, for Rhodoplanes (Hiraishi and Ueda, 1994), which indicates that they can utilize a vast range of organic compounds as a C source. In spite of the limited knowledge regarding the physiological abilities of these microorganisms, they are known to acquire C from different sources. Their effective mechanisms regarding C utilization may be advantageous for their establishment and survival in recently deglaciated mineral debris. Based on our data, we cannot unequivocally determine which endogenous source dominates the pool of microbial pioneers colonizing recently exposed soils. We assume that pioneering bacteria originating from sub- and supraglacial sediments were transported with water flow formed during glacier melting, deposited on newly exposed soils, and therefore contributed to the colonization of recently deglaciated terrains.
A few fungal indicator OTUs were shared among habitats. Nevertheless, basidiomycete OTUs of the yeast-like genera Leucosporidium and Rhodotorula were associated with snow and supraglacial sediments. Co-correlation patterns of indicator OTUs affiliated to Basidiomycota show that fungi present in snow shared the same habitat as those present in supraglacial sediments, suggesting that fungi present in snow may also survive in supraglacial sediments. Interestingly, these habitats were not spatially linked (Supplementary Table S1) although supraglacial sediments may originate from atmospheric material deposited during snow melt, which would explain their similarity in fungal communities. This finding indicates that environmental filtering may dominate the establishment of fungal communities in the Damma glacier catchment. Yeast-like Basidiomycota are found in the cryosphere across the globe because of structural and functional adaptations to the cold, such as increased membrane fluidity and production of cryoprotectants (Buzzini et al., 2012). Although snow and supraglacial sediments are exposed to light and may experience large temperature fluctuations during the growing season, these environments remain relatively cold because of the proximity of glacier ice. We cannot, however, exclude the possibility that other factors, such as paucity of C and N, as well as high UV exposure, may favour extremophile yeasts in these habitats (Gostinčar et al., 2010, 2012). In the other habitats investigated, fungal indicator OTUs were mostly affiliated to genera known to be saprotrophic, such as Cladophialophora, Cadophora and Omphalina (Clemmensen et al., 2013). Interestingly, fungal indicator OTUs characterizing subglacial sediments, ice and barren soils consisted of plant pathogens (for example, Heterobasidion and Peniophora), ericoid (Rhizoscyphus) and arbuscular (Entrophospora) mycorrhizae (Clemmensen et al., 2013). Although we could not exclude the possibility that these plant-associated fungi were active, they were probably attached to plant or root particles trapped in ice and recently released with glacier melting.
Despite the high-resolution description of bacterial and fungal diversity in various habitats (soils, endo- and exogenous sources of microorganisms), our approach failed to provide useful information about the archaeal communities. Archaeal OTUs could be detected in all habitats investigated but most of these OTUs were unclassified at the phylum level (>94% of the sequence number). Several studies targeting the hypervariable region V3–V5 of the archaeal 16S ribosomal marker demonstrated such a caveat (La Duc et al., 2012; Klindworth et al., 2013; Park et al., 2014). Archaea are known to occur in the Damma glacier forefield where their α-diversity is low in barren soils (Zumsteg et al., 2012). Moreover, Lazzaro et al. (2015) failed to identify them in snow, whereas we could amplify archaeal fragments in snow samples. Similar to results reported by Hamilton et al. (2013), we found archaeal OTUs in subglacial sediments and basal ice. However, given the low classification percentage of archaeal reads, robust conclusions cannot be drawn confidently concerning the archaeal community in the habitats investigated. We suggest that archaea in our ecosystem only comprised a minor part of the soil microbiota, as demonstrated in a recent metagenomics study of an alpine glacial system (Edwards et al., 2013a). Qian et al. (2011) also observed poor classification success at the kingdom level of archaeal communities in the Red Sea and further suggested that this finding may arise from an underrepresentation of archaeal sequences in databases and unspecific primers. Our findings further suggest that this kingdom should be extensively investigated in future studies in order to document adequately changes in archaeal diversity.
The origin of the C and nutrients necessary to allow the establishment of microbial communities in recently deglaciated mineral debris is a critical unknown and the subject of ongoing debate (Bardgett et al., 2007; Freeman et al., 2009; Mladenov et al., 2012; Bradley et al., 2014). Other studies have shown that atmospheric deposition (snow, rain and aeolian dust) represents sources of C and nutrients to terrestrial ecosystems (Waldrop et al., 2004; Brankatschk et al., 2011; Wortman et al., 2012). We found that concentrations of DOC, DON, ammonium, nitrate, phosphate and sulphate in deposition were higher than in ice and glacier stream indicating that atmospheric deposition may significantly add C, N and other nutrients to barren nutrient-depleted soils (Brankatschk et al., 2011; Reay et al., 2008; Mladenov et al., 2012; Stres et al., 2013). In addition, the differences in DOC, DON and nutrient concentrations were correlated with changes in bacterial and fungal community structures, as well as with the relative abundances of particular OTUs. For example, we found that several bacterial indicator OTUs, such as Bradyrhizobium, Gemmata, Phormidium and Propionicimonas, were correlated with changes in DOC. These microorganisms, which were mainly associated with barren soils, harbour diverse strategies to gain energy and carbon. Isolates affiliated to Phormidium and Bradyrhizobium genera consist of phototrophs (Swingley et al., 2009; Janatková et al., 2013) and could thus provide autochthonous sources of C to heterotrophs present in barren soils, such as OTUs related to Gemmata and Propionicimonas known to thrive on diverse C sources (Ward et al., 2006; Akasaka, 2003).
In conclusion, this is the first comprehensive study simultaneously investigating bacterial and fungal diversity of glacial (ice, glacier stream, sub- and supraglacial sediments) and proglacial habitats (barren soils) as well as atmospheric deposition (snow, rain and aeolian dust) in the central Swiss Alps. Although bacterial communities present in proglacial barren soils resembled those in ice, glacier stream, and sub- and supraglacial sediments, fungal communities differed more distinctly among habitats. This observation suggests that endogenous sources of bacteria are the most dominant colonizers of mineral debris released during glacier retreat. Most of these bacteria were from genera known to thrive under harsh conditions (UV exposure and limitations of C and nutrients) and to utilize various organic compounds as a C source. In contrast, most of the fungal indicator OTUs were distinct among habitats, suggesting that fungal dispersal is limited across the atmospheric, glacier and proglacial environments. Nonetheless, yeasts putatively adapted to cold habitats were associated with snow and supraglacial sediments pointing to potential environmental filtering favouring these fungi in cold habitats of the Damma glacier catchment. Overall, our research indicates that atmospheric deposition (aeolian dust, rain and snow) may have a crucial role in the development of recently formed terrestrial ecosystems by providing available sources of energy, carbon and nutrients rather than by fuelling the pools of microbial pioneers. With the rapid disappearance of most glacier ecosystems worldwide, it becomes increasingly important to understand the biological dynamics of these environments in order to anticipate potential ecological trajectories in a warming world.
References
Akasaka H . (2003). Propionicimonas paludicola gen. nov., sp. nov., a novel facultatively anaerobic, Gram-positive, propionate-producing bacterium isolated from plant residue in irrigated rice-field soil. Int J Syst Evol Microbiol 53: 1991–1998.
Amato P, Hennebelle R, Magand O, Sancelme M, Delort A-M, Barbante C et al. (2007). Bacterial characterization of the snow cover at Spitzberg, Svalbard. FEMS Microbiol Ecol 59: 255–264.
Amato P, Ménager M, Sancelme M, Laj P, Mailhot G, Delort A-M . (2005). Microbial population in cloud water at the Puy de Dôme: implications for the chemistry of clouds. Atmos Environ 39: 4143–4153.
Anesio AM, Hodson AJ, Fritz A, Psenner R, Sattler B . (2009). High microbial activity on glaciers: importance to the global carbon cycle. Glob Chang Biol 15: 955–960.
Anesio AM, Sattler B, Foreman C, Telling J, Hodson A, Tranter M et al. (2010). Carbon fluxes through bacterial communities on glacier surfaces. Ann Glaciol 51: 32–40.
Bajerski F, Wagner D . (2013). Bacterial succession in Antarctic soils of two glacier forefields on Larsemann Hills, East Antarctica. FEMS Microbiol Ecol 85: 128–142.
Barberán A, Henley J, Fierer N, Casamayor EO . (2014). Structure, inter-annual recurrence, and global-scale connectivity of airborne microbial communities. Sci Total Environ 487: 187–195.
Bardgett RD, Richter A, Bol R, Garnett MH, Bäumler R, Xu X et al. (2007). Heterotrophic microbial communities use ancient carbon following glacial retreat. Biol Lett 3: 487–490.
Bernasconi SM, Bauder A, Bourdon B, Brunner I, Bünemann E, Chris I et al. (2011). Chemical and biological gradients along the Damma glacier soil chronosequence, Switzerland. Vadose Zo J 10: 867.
Bowers RM, Lauber CL, Wiedinmyer C, Hamady M, Hallar AG, Fall R et al. (2009). Characterization of airborne microbial communities at a high-elevation site and their potential to act as atmospheric ice nuclei. Appl Environ Microbiol 75: 5121–5130.
Bowers RM, McCubbin IB, Hallar AG, Fierer N . (2012). Seasonal variability in airborne bacterial communities at a high-elevation site. Atmos Environ 50: 41–49.
Bradley J, Singarayer J, Anesio A . (2014). Microbial community dynamics in the forefield of glaciers. Proc R Soc B Biol Sci 281: 20140882.
Brankatschk R, Töwe S, Kleineidam K, Schloter M, Zeyer J . (2011). Abundances and potential activities of nitrogen cycling microbial communities along a chronosequence of a glacier forefield. ISME J 5: 1025–1037.
Brown SP, Jumpponen A . (2014). Contrasting primary successional trajectories of fungi and bacteria in retreating glacier soils. Mol Ecol 23: 481–497.
Brunner I, Plötze M, Rieder S, Zumsteg A, Furrer G, Frey B . (2011). Pioneering fungi from the Damma glacier forefield in the Swiss Alps can promote granite weathering. Geobiology 9: 266–279.
Buzzini P, Branda E, Goretti M, Turchetti B . (2012). Psychrophilic yeasts from worldwide glacial habitats: diversity, adaptation strategies and biotechnological potential. FEMS Microbiol Ecol 82: 217–241.
De Cáceres M, Legendre P, Moretti M . (2010). Improving indicator species analysis by combining groups of sites. Oikos 119: 1674–1684.
Cameron KA, Hagedorn B, Dieser M, Christner BC, Choquette K, Sletten R et al. (2015). Diversity and potential sources of microbiota associated with snow on western portions of the Greenland ice sheet. Environ Microbiol 17: 594–609.
Chuvochina MS, Marie D, Chevaillier S, Petit J-R, Normand P, Alekhina I A et al. (2011). Community variability of bacteria in alpine snow (Mont Blanc) containing Saharan dust deposition and their snow colonisation potential. Microbes Environ 26: 237–247.
Clarke K, Gorley R . (2006). PRIMER v6: User Manual/Tutorial. PRIMER-E. Plymouth.
Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H et al. (2013). Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 339: 1615–1618.
Collaud Coen M, Weingartner E, Schaub D, Hueglin C, Corrigan C, Schwikowski M et al. (2003). Saharan dust events at the Jungfraujoch: detection by wavelength dependence of the single scattering albedo and analysis of the events during the years 2001 and 2002. Atmos Chem Phys Discuss 3: 5547–5594.
Dabney A, Storney JD . (2014). qvalue: Q-value estimation for false discovery rate control. R package version 1.38.0.
Darcy JL, Lynch RC, King AJ, Robeson MS, Schmidt SK . (2011). Global distribution of Polaromonas phylotypes-evidence for a highly successful dispersal capacity. PLoS One 6: e23742.
DeLeon-Rodriguez N, Lathem TL, Rodriguez-R LM, Barazesh JM, Anderson BE, Beyersdorf AJ et al. (2013). Microbiome of the upper troposphere: species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc Natl Acad Sci USA 110: 2575–2580.
De Mendiburu F . (2012). agricolae: Statistical procedures for agricultural research. R package version 1-0.3.
Dorfman R . (1979). A formula for the Gini coefficient. Rev Econ Stat 61: 146–149.
La Duc MT, Vaishampayan P, Nilsson HR, Torok T, Venkateswaran K . (2012). Pyrosequencing-derived bacterial, archaeal, and fungal diversity of spacecraft hardware destined for Mars. Appl Environ Microbiol 78: 5912–5922.
Edwards A, Douglas B, Anesio AM, Rassner SM, Irvine-Fynn TDL, Sattler B et al. (2013b). A distinctive fungal community inhabiting cryoconite holes on glaciers in Svalbard. Fungal Ecol 6: 168–176.
Edwards A, Irvine-Fynn T, Mitchell AC, Rassner SME . (2014). A germ theory for glacial systems? Wiley Interdiscip Rev Water 1: 331–340.
Edwards A, Rassner S, Anesio AM . (2013b). Contrasts between the cryoconite and ice-marginal bacterial communities of Svalbard glaciers. Polar Res 32: 1–9.
Favet J, Lapanje A, Giongo A, Kennedy S, Aung Y-Y, Cattaneo A et al. (2013). Microbial hitchhikers on intercontinental dust: catching a lift in Chad. ISME J 7: 850–867.
Felip M, Sattler B . (1995). Highly active microbial communities in the ice and snow cover of high mountain lakes. Appl Environ Microbiol 61: 2394–2401.
Franzetti A, Tatangelo V, Gandolfi I, Bertolini V, Bestetti G, Diolaiuti G et al. (2013). Bacterial community structure on two alpine debris-covered glaciers and biogeography of Polaromonas phylotypes. ISME J 7: 1483–1492.
Freeman KR, Pescador MY, Reed SC, Costello EK, Robeson MS, Schmidt SK . (2009). Soil CO2 flux and photoautotrophic community composition in high-elevation, ‘barren’ soil. Environ Microbiol 11: 674–686.
Frey B, Bühler L, Schmutz S, Zumsteg A, Furrer G . (2013). Molecular characterization of phototrophic microorganisms in the forefield of a receding glacier in the Swiss Alps. Environ Res Lett 8: 015033.
Frey B, Niklaus PA, Kremer J, Lüscher P, Zimmermann S . (2011). Heavy-machinery traffic impacts methane emissions as well as methanogen abundance and community structure in oxic forest soils. Appl Environ Microbiol 77: 6060–6068.
Frey B, Rieder SR, Brunner I, Plötze M, Koetzsch S, Lapanje A et al. (2010). Weathering-associated bacteria from the Damma Glacier forefield: physiological capabilities and impact on granite dissolution. Appl Environ Microbiol 76: 4788–4796.
Gobet A, Quince C, Ramette A . (2010). Multivariate cutoff level analysis (MultiCoLA) of large community data sets. Nucleic Acids Res 38: e155.
Gostinčar C, Grube M, de Hoog S, Zalar P, Gunde-Cimerman N . (2010). Extremotolerance in fungi: evolution on the edge. FEMS Microbiol Ecol 71: 2–11.
Gostinčar C, Muggia L, Grube M . (2012). Polyextremotolerant black fungi: oligotrophism, adaptive potential, and a link to lichen symbioses. Front Microbiol 3: 390.
Hamilton TL, Peters JW, Skidmore ML, Boyd ES . (2013). Molecular evidence for an active endogenous microbiome beneath glacial ice. ISME J 7: 1402–1412.
Hartmann M, Frey B, Mayer J, Mäder P, Widmer F . (2015). Distinct soil microbial diversity under long-term organic and conventional farming. ISME J 9: 1177–1194.
Hartmann M, Howes CG, VanInsberghe D, Yu H, Bachar D, Christen R et al. (2012). Significant and persistent impact of timber harvesting on soil microbial communities in Northern coniferous forests. ISME J 6: 2199–2218.
Hartmann M, Niklaus PA, Zimmermann S, Schmutz S, Kremer J, Abarenkov K et al. (2014). Resistance and resilience of the forest soil microbiome to logging-associated compaction. ISME J 8: 226–244.
Hell K, Edwards A, Zarsky J, Podmirseg SM, Girdwood S, Pachebat JA et al. (2013). The dynamic bacterial communities of a melting high Arctic glacier snowpack. ISME J 7: 1814–1826.
Hervàs A, Camarero L, Reche I, Casamayor EO . (2009). Viability and potential for immigration of airborne bacteria from Africa that reach high mountain lakes in Europe. Environ Microbiol 11: 1612–1623.
Hiraishi A, Ueda Y . (1994). Rhodoplanes gen. nov., a new genus of phototrophic bacteria including Rhodopseudomonas rosea as Rhodoplanes roseus comb. nov. and Rhodoplanes elegans sp. nov. Int J Syst Bacteriol 44: 665–673.
Hodson A, Anesio A, Tranter M . (2008). Glacial ecosystems. Ecol Stud 78: 41–67.
Holm NC, Gliesche CG, Hirsch P . (1996). Diversity and structure of hyphomicrobium populations in a sewage treatment plant and its adjacent receiving lake. Appl Environ Microbiol 62: 522–528.
Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM . (2001). Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 67: 4399–4406.
Incagnone G, Marrone F, Barone R, Robba L, Naselli-Flores L . (2014). How do freshwater organisms cross the ‘dry ocean’? A review on passive dispersal and colonization processes with a special focus on temporary ponds. Hydrobiologia, 1–21.
Janatková K, Reháková K, Doležal J, Simek M, Chlumská Z, Dvorský M et al. (2013). Community structure of soil phototrophs along environmental gradients in arid Himalaya. Environ Microbiol 15: 2505–2516.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M et al. (2013). Evaluation of general 16 S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41: e1.
Larose C, Dommergue A, Vogel TM . (2013). The dynamic arctic snow pack: an unexplored environment for microbial diversity and activity. Biology 2: 317–330.
Lazzaro A, Wismer A, Schneebeli M, Erny I, Zeyer J . (2015). Microbial abundance and community structure in a melting alpine snowpack. Extremophiles 19: 631–642.
Legendre P, Anderson M . (1999). Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69: 1–24.
Letunic I, Bork P . (2011). Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39: W475–W478.
Lindström ES, Langenheder S . (2012). Local and regional factors influencing bacterial community assembly. Env Microbiol Rep 4: 1–9.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Menon M, Rousseva S, Nikolaidis NP, van Gaans P, Panagos P, de Souza DM et al. (2014). SoilTrEC: a global initiative on critical zone research and integration. Environ Sci Pollut Res 21: 3191–3195.
Mladenov N, Williams MW, Schmidt SK, Cawley K . (2012). Atmospheric deposition as a source of carbon and nutrients to barren, alpine soils of the Colorado Rocky Mountains. Biogeosciences 9: 2375–2424.
Musilova M, Tranter M, Bennett SA, Wadham J, Anesio AM . (2015). Stable microbial community composition on the Greenland Ice Sheet. Front Microbiol 6: 1–10.
Naff CS, Darcy JL, Schmidt SK . (2013). Phylogeny and biogeography of an uncultured clade of snow chytrids. Environ Microbiol 15: 2672–2680.
Nakatsu CH, Hristova K, Hanada S, Meng X-Y, Hanson JR, Scow KM et al. (2006). Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int J Syst Evol Microbiol 56: 983–989.
Naselli-Flores L, Termine R, Barone R . (2016). Phytoplankton colonization patterns. Is species richness depending on distance among freshwaters and on their connectivity? Hydrobiologia 764: 103–113.
Oksanen J, Guillaume Blanchet F, Kindt R, Legendre P, Minchin PR, O’Hara RB et al. (2012). vegan: Community Ecology Package. R Package version 2.0-3.
Park T-J, Ding W, Cheng S, Brar MS, Ma APY, Tun HM et al. (2014). Microbial community in microbial fuel cell (MFC) medium and effluent enriched with purple photosynthetic bacterium (Rhodopseudomonas sp.). AMB Express 4: 22.
Pearce DA, Bridge PD, Hughes KA, Sattler B, Psenner R, Russell NJ . (2009). Microorganisms in the atmosphere over Antarctica. FEMS Microbiol Ecol 69: 143–157.
Priscu J, Christner B . (2004). Earth’s icy biosphere. In: Microbial Diversity and Prospecting. Bull A (ed) American Society for Microbiology: Washington, DC, pp 130–144.
Qian P-Y, Wang Y, Lee OO, Lau SCK, Yang J, Lafi FF et al. (2011). Vertical stratification of microbial communities in the Red Sea revealed by 16S rDNA pyrosequencing. ISME J 5: 507–518.
R development Core Team. (2012). R: A language and environment for statistical Computing http://www.r-project.org/.
Reay DS, Dentener F, Smith P, Grace J, Feely RA . (2008). Global nitrogen deposition and carbon sinks. Nat Geosci 1: 430–437.
Řeháková K, Stibal M, Šabacká M, Řehák J . (2010). Survival and colonisation potential of photoautotrophic microorganisms within a glacierised catchment on Svalbard, High Arctic. Polar Biol 33: 737–745.
Revelle W . (2014). psych: Procedures for psychological, psychometric, and personality research. R package version 1.4.8.
Rime T, Hartmann M, Brunner I, Widmer F, Zeyer J, Frey B . (2015). Vertical distribution of the soil microbiota along a successional gradient in a glacier forefield. Mol Ecol 24: 1091–1108.
Šantl-Temkiv T, Finster K, Hansen BM, Nielsen NW, Karlson UG . (2012). The microbial diversity of a storm cloud as assessed by hailstones. FEMS Microbiol Ecol 81: 684–695.
Šantl-Temkiv T, Finster K, Hansen BM, Pašić L, Karlson UG . (2013). Viable methanotrophic bacteria enriched from air and rain can oxidize methane at cloud-like conditions. Aerobiologia (Bologna) 29: 373–384.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Schmidt S, Nemergut D, Darcy J, Lynch R . (2014). Do bacterial and fungal communities assemble differently during primary succession? Mol Ecol 23: 254–258.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504.
Smittenberg RH, Gierga M, Göransson H, Christl I, Farinotti D, Bernasconi SM . (2012). Climate-sensitive ecosystem carbon dynamics along the soil chronosequence of the Damma glacier forefield, Switzerland. Glob Chang Biol 18: 1941–1955.
Spearman C . (1904). The proof and measurement of association between two things. Am J Psychol 15: 72–101.
Stackebrandt E, Verbarg S, Frühling A, Busse H-J, Tindall BJ . (2009). Dissection of the genus Methylibium: reclassification of Methylibium fulvum as Rhizobacter fulvus comb. nov., Methylibium aquaticum as Piscinibacter aquaticus gen. nov., comb. nov. and Methylibium subsaxonicum as Rivibacter subsaxonicus gen. nov., comb. n. Int J Syst Evol Microbiol 59: 2552–2560.
Stres B, Sul W, Murovec B, Tiedje J . (2013). Recently deglaciated high-altitude soils of the Himalaya: diverse environments, heterogenous bacterial communities and long-range dust inputs from the upper troposphere. PLoS One 8: e76640.
Swingley WD, Blankenship RE, Raymond J . (2009) Evolutionary relationships among purple photosynthetiv bacteria and origin of proteobacterial photosynthetic systems. In The Purple Phototrophic Bacteria, Hunter N, Daldal F, Thurnauer MC, Thomas BJ (eds). Springer: The Netherlands, pp 17–29.
Symons CC, Arnott SE . (2014). Timing is everything: priority effects alter community invasibility after disturbance. Ecol Evol 4: 397–407.
Tedersoo L, Bahram M, Toots M, Diédhiou AG, Henkel TW, Kjøller R et al. (2012). Towards global patterns in the diversity and community structure of ectomycorrhizal fungi. Mol Ecol 21: 4160–4170.
Vaïtilingom M, Amato P, Sancelme M, Laj P, Leriche M, Delort A-M . (2010). Contribution of microbial activity to carbon chemistry in clouds. Appl Environ Microbiol 76: 23–29.
Vorholt JA . (2012). Microbial life in the phyllosphere. Nat Rev Microbiol 10: 828–840.
Waldrop MP, Zak DR, Sinsabaugh RL, Gallo M, Lauber C . (2004). Nitrogen deposition modifies soil carbon storage through changes in microbial enzymatic activity. Ecol Appl 14: 1172–1177.
Ward N, Staley JT, Fuerst JA, Giovannoni S . (2006). The order Planctomycetales, including the genera Planctomyces, Pirellula, Gemmata and Isosphaera and the Candidatus Genera Brocadia, Kuenenia and Scalindua. Prokaryotes 7: 757–793.
Werner GDA, Kries ET . (2015). Order of arrival structures arbuscular mycorrhizal colonization of plants. New Phytol 205: 1515–1524.
Wickham H . (2009) ggplot2: Elegant Graphics for Data Analysis. Springer: New York, http://had.co.nz/ggplot2/book.
Wilhelm L, Singer GA, Fasching C, Battin TJ, Besemer K . (2013). Microbial biodiversity in glacier-fed streams. ISME J 7: 1651–1660.
Womack AM, Bohannan BJM, Green JL . (2010). Biodiversity and biogeography of the atmosphere. Philos Trans R Soc Lond B Biol Sci 365: 3645–3653.
Wortman E, Tomaszewski T, Waldner P, Schleppi P, Thimonier A, Eugster W et al. (2012). Atmospheric nitrogen deposition and canopy retention influences on photosynthetic performance at two high nitrogen deposition Swiss forests. Tellus B 64: 17216.
Zhang S, Yang G, Wang Y, Hou S . (2010). Abundance and community of snow bacteria from three glaciers in the Tibetan Plateau. J Environ Sci 22: 1418–1424.
Zumsteg A, Luster J, Göransson H, Smittenberg RH, Brunner I, Bernasconi SM et al. (2012). Bacterial, archaeal and fungal succession in the forefield of a receding glacier. Microb Ecol 63: 552–564.
Acknowledgements
This study was funded by the Swiss National Science Foundation (SNSF) under the grant number 31003A-138321. We gratefully thank the Central Laboratory, Noureddine Hajjar and Beat Stierli (Swiss Federal Research Institute WSL) for completing chemical analyses and providing help in the laboratory. We thank Professor Dr Josef Zeyer (ETH Zürich) and Dr Melissa Dawes (Swiss Federal Research Institute WSL) for their valuable comments during the writing of this article. We acknowledge the Genetic Diversity Center (GDC) at ETH Zürich and the contribution of scientists at the McGill University and Génome Québec Innovation Center, Montréal, Canada, for conducting the 454-pyrosequencing. We confirm that the material presented in the manuscript is original research that has not been previously published and submitted for publication elsewhere.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Rime, T., Hartmann, M. & Frey, B. Potential sources of microbial colonizers in an initial soil ecosystem after retreat of an alpine glacier. ISME J 10, 1625–1641 (2016). https://doi.org/10.1038/ismej.2015.238
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.238
This article is cited by
-
Ecological significance of Candidatus ARS69 and Gemmatimonadota in the Arctic glacier foreland ecosystems
Applied Microbiology and Biotechnology (2024)
-
Microbial Community Structure and Metabolic Potential at the Initial Stage of Soil Development of the Glacial Forefields in Svalbard
Microbial Ecology (2023)
-
Unexpected high carbon losses in a continental glacier foreland on the Tibetan Plateau
ISME Communications (2022)
-
Microdiversity characterizes prevalent phylogenetic clades in the glacier-fed stream microbiome
The ISME Journal (2022)
-
Routes and rates of bacterial dispersal impact surface soil microbiome composition and functioning
The ISME Journal (2022)
