
Abstract
The prevalence of obesity is increasing alarmingly to epidemic proportions in children and adolescents, especially in industrialized countries. The finding that overweight children, especially girls, tend to mature earlier than lean children has led to the hypothesis that the degree of body fatness may trigger the neuroendocrine events that lead to the onset of puberty. Obese children have high leptin levels, and these may play a role in their earlier onset of puberty. Leptin receptors have been identified in the hypothalamus, gonadotrope cells of the anterior pituitary, and ovarian follicular cells, as well as Leydig cells. Leptin accelerates gonadotropin-releasing hormone (GnRH) pulsatility in hypothalamic neurons, and it has a direct effect on the anterior pituitary. Leptin administration at low doses may have a permissive, threshold effect on the central networks that regulate gonadotropin secretion. However, at high levels, such as those in obese people, it can have an inhibitory effect on the gonads. Children with obesity also have increased adrenal androgen levels, which may be involved in the accelerated growth of these children before puberty. Recent data indicate that leptin has a specific role in stimulating the activity of enzymes essential for the synthesis of adrenal androgens. Children with exogenous obesity frequently show an increase in height velocity with tall stature for age despite low growth hormone levels. Our group has shown that leptin acts as a skeletal growth factor, with a direct effect on skeletal growth centers, in the mice mandibular condyle, a model of endochondral ossification. In summary, obesity is associated with early puberty. Elevated leptin levels might have a permissive effect on the pubertal process and pubertal growth.
Similar content being viewed by others

Introduction
Obesity is a major public health problem that has grown to epidemic proportions throughout the world.1,2 Rates in adults range from 10 to 25% in most Western European countries and from 20 to 25% in some parts of the United States. In children and adolescents, the National Health and Nutrition Examination Survey (NHANES III) reported an estimated 20% prevalence of obesity, defined as a body mass index (BMI) greater than the 85th percentile.1 For all age groups and degrees of obesity, girls are at higher risk than boys.
Obesity is not restricted to a single ethnic, age, or socioeconomic group, although cultural, environmental, and genetic factors clearly play a role.1 The environmental factors that contribute to the development of a high degree of body fatness early in life include a high proportion of sedentary activities (eg, TV viewing), low proportion of physical activity, and a shift in diet towards more fast foods with high fat and calorie content. Among the genetic factors, polymorphism or mutations in any of the following genes may be involved in the pathophysiology of obesity: β-adrenergic receptor, tumor necrosis factor (TNF), pro-opiomelanocortin (POMC), neuropeptide Y (NPY), NPY-receptor, melanocortin receptor (MC4R), leptin, and leptin receptor.3
Severe obesity is associated with a higher likelihood of continued overweight. This risk is much higher in children with obese parents. Studies have shown that parental obesity is the most important risk factor of obesity in children,4,5 owing to both the genetic influence and the shared environment (lifestyle, nutritional habits, food preferences, etc).
Among the most common sequelae of childhood obesity are hypertension and dyslipidemia, with increased risk of coronary heart disease, type II diabetes, respiratory problems, pseudotumor cerebri, and orthopedic and psychosocial problems. Since 85% of obese youngsters will become obese adults, the comorbidity exerts an important toll on industrialized societies.
Several critical periods in childhood have been identified in the development and persistence of obesity.6 In the gestational period, there is a direct association of high birth weight with subsequent adiposity.7 Thereafter, in the first year of life, children undergo changes in nutritional behavior which may influence adiposity in later life. Another crucial period is ages of 5-8 years, where adiposity increases after its nadir in childhood (‘adiposity rebound’). An early adiposity rebound may serve an index of further obesity.8,9 The final risk period for the development of persistent obesity is adolescence.10
Leptin is a protein product of the obesity (ob) gene. It is secreted as a hormone mainly from white adipose tissue and serves as a signal for the brain of the body's energy stores.11 By reducing food intake and increasing thermogenesis, leptin controls body fat tissue and, hence, body weight.12,13 Studies of its physiologic action in humans have shown a strong positive correlation between serum leptin concentrations and the percentage of body fat.14,15
Puberty in mammals is physiologically gated by the body's energy resources. According to the classic studies of Kennedy and Mitra16 and the work of Frisch et al,17 the timing of sexual maturation is associated with body weight and composition. That is, accelerated linear growth and reproductive maturity are affected by peripheral metabolic factors, which signal the body size and fat content. Before puberty can occur, a critical body weight or fat mass must be achieved.18 As such, alterations in diet and exercise exert a powerful influence on pubertal maturation.19,20 The observation that overweight girls tend to mature earlier than lean girls21 has led to the hypothesis that the degree of body fatness may trigger the neuroendocrine events that lead to the onset of menses.22 Kaplowitz et al23 reported that obesity, as measured by BMI, is significantly associated with early puberty in white girls and to a lesser extent in black girls. A correlation was also found between age at adiposity rebound and age at menarche.24 As obese children have higher leptin levels than lean children, this may be an important factor in their earlier onset of puberty.
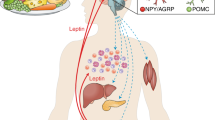
The present study reviews the findings clearly implicating the leptin hormone in the pathophysiology of obesity. Specifically, it focuses on the complicated interaction between obesity, leptin, the pubertal process, and the pubertal growth spurt (Figure 1).

Leptin function at various target tissues.
Obesity and puberty
Normal puberty consists of two distinct processes: maturation of gonadal function, known as gonadarche, and increased adrenal androgen secretion, known as adrenarche.
Role of leptin in gonadarche (hypothalmic-pituitary-gonadal (HPG) axis)
The medial basal hypothalamus contains a pulse generator that is responsible for the onset of the episodic secretion of hypothalamic gonadotropin-releasing hormone (GnRH), which in turn, results in the pulsatile secretion of the pituitary gonadotropins, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) during puberty. LH stimulates Leydig cell secretion of testosterone in boys, but has little effect in girls until after ovulation occurs. FSH stimulates follicle formation and estrogen secretion in girls, with little effect in boys until spermarche.
Leptin receptors (ob-Rs) have been identified in the hypothalamus and in the gonadotrope cells of the anterior pituitary.25 In the hypothalamus, leptin has a direct stimulatory effect on the HPG axis by accelerating GnRH secretion (but not pulse amplitude) in the arcuate hypothalamic neurons in a dose-dependent manner.26 In the anterior pituitary, leptin directly stimulates the release of LH and, to a lesser extent, FSH via nitric oxide synthase activation in the gonadotropes.27 The endocrine and direct paracrine effects of leptin on the gonads are implied by the expression of functional leptin receptors on the surface of ovarian follicular cells, including granulose, theca, and interstitial cells,28 as well as Leydig cells.29
Other downstream effectors of leptin are important in the control of feeding, but their influence on puberty is less well defined. NPY, a potent stimulator of food intake, has an inhibitory effect on GnRH secretion.30 Leptin administration has been shown to decrease the expression of NPY in the arcuate nucleus, and consequently to remove the inhibitory action of NPY on pulsatile GnRH release.31
Leptin levels exhibit significant changes during progressive pubertal stages, with a distinct dimorphism between boys and girls. In boys, there is a prepubertal peak of serum leptin levels preceding the rise of free testosterone, growth hormone (GH), and insulin-like growth factor (IGF-1). Thereafter, about 3 y after the rise in serum testosterone levels, leptin levels fall to baseline concentrations.32 In girls, leptin levels rise throughout puberty, concomitant with the rise in estrogen levels.32 Even after correcting for body weight and fat mass, females have higher serum leptin levels than males.33 This sexual dimorphism is related to several factors. First, the pulse amplitude of leptin secretion from adipose tissue is two- to three-fold higher in females than in males.34 Second, fat mass is increased in females who have a differential fat distribution, with a higher subcutaneous/visceral fat ratio than males (leptin mRNA expression is higher in subcutaneous than visceral fat).35 Third, females have higher total serum leptin levels and lower leptin-binding protein levels than males, indicating higher free leptin levels.36 Finally, female adipose tissue may be more sensitive to hormones that stimulate leptin production, such as glucocorticoids. Halleux et al37 demonstrated that physiological concentrations of glucocorticoids stimulated leptin secretion by enhancing the pretranslation process in human visceral and subcutaneous fat, especially in obese subjects.
Patients lacking leptin protein38 or functional leptin receptors39 do not attain pubertal maturity and have low serum FSH and LH. This finding suggests that hypogonadotropic hypogonadism is a feature of congenital leptin deficiency in humans. Farooqi et al40,41 reported that administration of recombinant leptin in a child with leptin deficiency restored pulsatile gonadotropin secretion.
At high physiologic doses, leptin appears to antagonize the augmenting effect of growth factors (IGF-1) and hormones (insulin, glucocorticoids) on gonadotropin-stimulated steroidogenesis in follicular and theca ovarian cells throughout the menstrual cycles42; in Leydig cells, it exerts inhibitory effects on testosterone production.29 Hence, leptin possesses a bimodal action on the HPG axis, depending on its serum levels. Specifically, leptin deficiency to the low levels seen in starvation or eating disorders results in HPG dysfunction, and leptin administration in low doses may have a permissive, threshold effect on the central networks that regulate gonadotropin secretion. By contrast, the high serum leptin levels seen in obese people may have an inhibitory effect on the gonads.
The findings of an earlier onset of the classic pubertal signs in female mice injected with leptin compared with controls43 suggested that leptin is the signal that transmits the information to the brain that fat stores are adequate to cover the energy requirements of reproduction. Leptin, therefore, may participate in the timing of puberty. Another recent study in female rats showed that leptin is able to reverse the delayed onset of puberty in animals receiving less than normal quantities of food.44 Thus, leptin is apparently not the signal that triggers the onset of puberty, but rather serves as a metabolic gate for puberty to progress.44 Palmert et al45 found that girls with central precocious puberty have slightly higher serum leptin levels than normal, healthy girls, even after correcting for BMI and Tanner stage. Furthermore, in a recent study of the relation between fat mass and age at menarche, researchers46 found that rising leptin levels during puberty were associated with a decline in the age at menarche46: Every 1 ng/ml increase in serum leptin levels was associated with a 1-month decrease in age at menarche, and every 1 kg increase in body fat was associated with a 13-day decrease in age at menarche. Apparently, there is a critical concentration or threshold of leptin for the timing of menarche. Leptin deficiency may be the primary reason for delayed puberty and menarche in individuals accustomed to absolute or relative dietary energy deficiency.
Soluble leptin receptor (sOB-R), the major leptin-binding protein in human circulation, may modulate leptin bioavailability and function. Kratzsch et al47 found inverse relations of sOB-R with age, pubertal stage, and body composition parameters, as well as with leptin concentrations. The increased sOB-R levels in subjects with hypoleptinemia may represent a mechanism to temporally preserve the reduced hormone levels in the circulation. Correlation analysis demonstrated that the parameters of growth and sexual maturation were more closely related to the ratio between leptin and sOB-R, free leptin index (FLI), than to leptin alone, predominantly in boys. Therefore, sOB-R levels may serve as another valuable tool to investigate the leptin axis during growth and sexual maturation.
In summary, leptin plays a dual role in gonadarche. First, it may be one of several factors that induce cellular maturation of the GnRH pulse generator. Maturation of the neuroendocrine networks may occur when fat stores are sufficient to maintain a threshold leptin concentration. Second, leptin may act as a signal of metabolic fuel availability to the GnRH pulse generator, such that the continuation of puberty in the adolescent is dependent on adequate leptin feedback from fat and muscle.
Role of obesity and leptin in adrenarche
According to cross-sectional observations, adrenarche begins about the same time as the preadolescent rise in BMI, gradual increase in plasma insulin, and increase in IGF-I serum levels.48 Remer and Manz49 reported a temporal proximity in individuals of the greatest increase in dehydroepiandrosterone (DHEA) sulfate (DHEAS) levels in early adrenarche with the greatest increase in BMI. This observation provided evidence that a change in nutritional status, measurable in the form of ΔBMI, is an important physiological regulator of adrenarche, regardless of individual adrenal androgen excretion level, age, and developmental stage. Although ΔBMI is associated with serum leptin levels, it may also be an integrated measure of the combined long-term actions of IGF-I and insulin, which have been shown to augment adrenal androgen production and the expression of steroidogenic enzymes in human adrenocortical cells.49 Adrenal androgen levels are increased in children with obesity50 and may therefore be responsible for their accelerated growth before puberty.51 This assumption is supported by data pointing to a specific, dose-dependent, stimulatory activity of leptin on 17-α-hydroxylase and 17–20 lyase, both enzymes essential for the synthesis of adrenal androgens.52
In view of these findings, we may assume that marked weight gain and obesity (associated with insulin resistance and high insulin levels) are probably involved in the development of premature adrenarche and the subsequent manifestations of functional ovarian hyperandrogenism and polycystic ovary syndrome. Cizza et al53 reported that girls with premature adrenarche had a higher BMI and a more than two-fold elevation in plasma leptin levels compared to nonadrenarchal, age-matched girls. They also had elevated levels of salivary and plasma cortisol, DHEA, DHEAS, androstenedione, estradiol, and esterone. The authors proposed that girls with premature adrenarche are characterized by features of increased adiposity and hypothalamic-pituitary-adrenal axis activity. In contrast, however, another study54 found that in patients with premature adrenarche, neither BMI nor leptin levels were correlated with the increased androgen levels.54
Prader–Willi syndrome (PWS) is the most frequent form of syndromal obesity. The phenotype is linked with a genetic hypothalamic defect. GH insufficiency and hypogonadism are described in PWS, and the gonadal development at puberty is typically delayed or incomplete. Surprisingly, however, premature adrenarche may be observed in affected children,55 eventhough their insulin and IGF-1 levels are below the normal average because of insufficient GH secretion.56 In addition, the mean adrenal androgen levels in patients with PWS are elevated above the normal range,57 like in healthy obese children. The typically delayed gonadal maturation in PWS may be contrasted by elevated androgen levels and premature adrenarche. These hormonal changes are amplified by obesity and related metabolic alteration, independent of the hypothalamic regulation.
Further research is needed to fully elucidate the role of increased leptin levels in these physiological and pathophysiological processes.
Role of obesity and leptin in pubertal growth
Growth in childhood depends on an intact and functioning GH-IGF-I axis. Androgens and estrogens do not contribute substantially to normal growth before the onset of puberty, but they play an important role in the pubertal growth spurt. In adolescents, the rising sex hormone levels may exert a growth-promoting effect via their stimulatory effect on the GH–IGF-I axis. However, in some clinical conditions, normal growth persists or is even accelerated in the presence of low GH serum levels. This has been observed in children with GH deficiency secondary to craniopharyngioma surgery. In these cases, the accelerated growth is associated with the development of marked obesity.58,59 Children with exogenous obesity also frequently show tall stature for age in association with an acceleration of epiphysial growth plate maturation.60 However, the linear growth often stops prematurely because of the advanced bone age, and in some cases, final height is below the adult height potential.
During the phase of normal or accelerated height velocity in obese children, plasma GH levels remain low, apparently as a result of both decreased GH secretion by the pituitary gland and increased GH clearance from plasma.61,62,63 Measurements performed during sleep and over 24 h have shown that GH secretion in obese children is reduced not only under physiological conditions, but also following pharmacologic stimulation with insulin, clonidine, arginine, levodopa, glucagon, GH-releasing hormone, and opiate peptides, much like in children with GH deficiency.64 Despite the reduction in circulating GH levels, obese children may have normal, increased or reduced, plasma IGF-I and GH-binding protein levels.65,66,67
The mechanism whereby obese children continue to grow despite the low levels of GH is not known. Several explanations have been postulated, such as obesity-induced hyperinsulinemia,68 hyperprolactinemia,68 induced bioactive but nonimmunoreactive GH molecules,69 and an increase in free IGF-I levels.67 The possibility that children with obesity may have an as yet unidentified ‘circulating factor’ that stimulates growth independent of the presence of GH has also been suggested.59
In an animal study, Maor et al70 reported on the presence of leptin receptors in cartilaginous skeletal growth centers involved in leptin-induced skeletal growth. Leptin is known to stimulate, in a dose-dependent manner, the width of the proliferative zone of the epiphysial growth plate and to increase the expression of chondroitin sulfate within the cartilaginous matrix. Thus, leptin induces both proliferation and differentiation of chondrocytes. Apparently, leptin acts as a skeletal growth factor with a direct peripheral effect on skeletal growth centers. Some of these effects on the growing bone may be mediated by the IGF system through an increase in local IGF-I receptor expression.
In humans, obesity is associated with central resistance to circulating leptin. A differential sensitivity between the center (hypothalamus) and periphery (epiphysial growth plate) to the effect of leptin might serve as an explanation for the accelerated growth in obese children.
Conclusions
The prevalence of obesity is increasing alarmingly among children and adolescents, especially in industrialized countries. Many of the metabolic and cardiovascular complications that are commonly associated with adult obesity begin in childhood. Among the most common sequelae of childhood obesity are hypertension, dyslipidemia, insulin resistance with increased risk of type II diabetes, and orthopedic and psychosocial problems (poor self-image, social isolation). However, physicians treating children and adolescents need to bear in mind that childhood obesity is associated with additional phenomena, namely, early puberty, premature adrenarche and the subsequent manifestation of polycystic ovary syndrome, and accelerated growth with impaired final height potential, all of which can already affect their quality of life. Understanding the antecedents of obesity-related complications in obese children is therefore of great importance.
References
Troiano F, Flegal K . Overweight children and adolescents: Description, epidemiology, and demographics. Pediatrics 1998; 101 (Suppl): 497–504.
Gortmaker SL, Dietz Jr WH, Sobol AM, Wehler CA . Increasing pediatric obesity in the United States. Am J Dis Child 1987; 141: 535–540.
Perusse L, Chagnon YC, Dionne FT, Bouchard C . The human obesity gene map: the 1996 update. Obesity Res 1997; 5: 49–61.
Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH . Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med 1997; 337: 869–873.
Maffeis C, Talamini G, Tato L . Influence of diet, physical activity, and parents' obesity on children's adiposity: A four-year longitudinal study. Int J Obes Relat Metab Disord 1998; 22: 758–764.
Dietz WH . Critical periods in childhood for the development of obesity. Am J Clin Nutr 1994; 59: 955–959.
Seidman DS, Laor A, Gale R, Stevenson DK, Danon YL . A longitudinal study of birth weight and being overweight in late adolescence. Am J Dis Child 1991; 145: 782–785.
Whitaker RC, Pepe MS, Wright JA, Seidel KD, Dietz WH . Early adiposity rebound and the risk of adult obesity. Pediatrics 1998; 101: E5.
Williams S, Davie G, Lam F . Predicting BMI in young adults from childhood data using two approaches to modeling adiposity rebound. Int J Obes Relat Metab Disord 1999; 23: 348–354.
Dietz WH . Health consequences of obesity in youth: childhood predictors of adult obesity. Pediatrics 1998; 101: 518–525.
Elmquist JK . Anatomic basis of leptin action in the hypothalamus. Front Horm Res 2000; 26: 21–41.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM . Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432.
Halaas JL, Gajiwala KS, Magherita M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Fredman JM . Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1996; 269: 543–546.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF . Serum immunoreactive leptin concentrations in normal-weight and obese humans. N Engl J Med 1996; 334: 292–295.
Hassink SG, Sheslow DV, deLancey E, Opentanova I, Considine RV, Caro JF . Serum leptin in children with obesity: relationship to gender and development. Pediatrics 1996; 98: 201–203.
Kennedy GC, Mitra J . Body weight and food intake as initiating factors for puberty in the rat. J Physiol 1963; 166: 408–418.
Frisch RE, Revelle R, Cook S . Components of weight at menarche and the initiation of the adolescent growth spurt in girls: estimation total water, lean body weight and fat. Hum Biol 1973; 45: 469–483.
Frisch RE . Pubertal adipose tissue: is it necessary for normal sexual maturation? Evidence from the rat and human female. Fed Proc 1980; 39: 2395–2400.
Frisch RE, Wyshak G, Vincent L . Delayed menarche and amenorrhea in ballet dancers. N Engl J Med 1980; 303: 17–19.
Warren MP . The effects of exercise on pubertal progression and reproductive functions in girls. J Clin Endocrinol Metab 1980; 51: 1150–1157.
Garn SM, Haskell JA . Fat and growth during childhood. Science 1959; 130: 1711–1712.
Frisch RE, McArthur JW . Menstrual cycles: fatness as a determinant of minimum weight for height necessary for their maintenance and onset. Science 1974; 185: 949–951.
Kaplowitz PB, Slora EJ, Wasserman RC, Pedlow SE, Heman-Giddens ME . Earlier onset of puberty in girls: relation to increased body mass index and race. Pediatrics 2001; 108: 347–353.
Williams S, Dickson N . Early growth, menarche, and adiposity rebound. Lancet 2002; 359: 580–581.
Jin L, Burguera BG, Couce ME, Scheithauer BW, Lamsan J, Eberhardt NL, Kulig E, Lloyd RV . Leptin and leptin receptor expression in normal and neoplastic human pituitary: evidence of a regulatory role for leptin on pituitary cell proliferation. J Clin Endocrinol Metab 1999; 84: 2903–2911.
Lebrethon MC, Vandersmissen E, Gerard A, Parent AS, Junien JL, Bourguignon JP . In vitro stimulation of the prepubertal rat gonadotropin-releasing-hormone pulse generator by leptin and neuropeptide Y through distinct mechanisms. Endocrinology 2000; 141: 1464–1469.
Yu WH, Kimura M, Walczewska A, Karanth S, McCann SM . Role of leptin in hypothalamic-pituitary function. Proc Natl Acad Sci USA 1997; 94: 1023–1028.
Karlsson C, Lindell K, Svensson E, Bergh C, Lind P, Billig H, Carlsson LM, Carlsson B . Expression of functional leptin receptors in the human ovary. J Clin Endocrinol Metab 1997; 82: 4144–4148.
Caprio M, Isidori AM, Carta AR, Moretti C, Dufau ML, Fabbri A . Expression of functional leptin receptors in rodent Leydig cells. Endocrinology 1999; 140: 4939–4947.
Plant TM, Shahab M . Neuroendocrine mechanisms that delay and initiate puberty in higher primates. Physiol Behav 2002; 77: 717–722.
El Majdoubi M, Sahu A, Ramaswamy S, Plant TM . Neuropeptide Y: a hypothalamic break restraining the onset of puberty in primates. Proc Natl Acad Sci USA 2000; 97: 6179–6184.
Clayton PE, Gill MS, Hall CM, Tillmann V, Whatmore AJ, Price DA . Serum leptin through childhood and adolescence. Clin Endocrinol (Oxf) 1997; 46: 727–733.
Saad MF, Damani S, Gingerich RL, Riad-Gabriel MG, Khan A, Boyadjian R, Jinagonda SD, el-Tawil K, Rude RK, Kamdar V . Sexual dimorphism in plasma leptin concentration. J Clin Endocrinol Metab 1997; 82: 579–584.
Licinio J, Negrao AB, Mantzoros C, Kaklamani V, Wong ML, Bongiorno PB, Negro PP, Mulla A, Veldhuis JD, Cearnal L, Flier JS, Gold PW . Sex differences in circulating human leptin pulse amplitude: clinical implications. J Clin Endocrinol Metab 1998; 83: 4140–4147.
Montague CT, Prins JB, Sanders L, Digby JE, O'Rahilly S . Depot- and sex-specific differences in human leptin mRNA expression: implications for the control of regional fat distribution. Diabetes 1997; 46: 342–347.
McConway MG, Johnson D, Kelly A, Griffin D, Smith J, Wallace AM . Difference in circulating concentrations of total, free and bound leptin relate to gender and body composition in adult humans. Ann Clin Biochem 2000; 37: 717–723.
Halleux CM, Servais I, Reul BA, Detry R, Brichard SM . Multihormonal control of ob gene expression and leptin secretion from cultured human visceral adipose tissue: increased responsiveness to glucocorticoids in obesity. J Clin Endocrinol Metab 1998; 83: 902–910.
Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD . A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet 1998; 18: 213–215.
Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougeneres P, Lebouc Y, Froguel P, Guy-Grand B . A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998; 392: 398–401.
Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA O'Rahilly S . Effect of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 1999; 341: 879–884.
Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O'Rahilly S . Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 2002; 110: 1093–1103.
Barkan D, Jia H, Dantes A, Vardimon L, Amsterdam A, Rubinstein M . Leptin modulates the glucocorticoid-induced ovarian steroidogenesis. Endocrinology 1999; 140: 1731–1738.
Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS . Leptin accelerates the onset of puberty in normal female mice. J Clin Invest 1997; 99: 391–395.
Cheung CC, Thornton JE, Kuijper JL, Weigle DS, Clifton DK, Steiner RA . Leptin is a metabolic gate for the onset of puberty in the female rat. Endocrinology 1997; 138: 855–858.
Palmert MR, Radovick S, Boepple P . Leptin levels in children with central precocious puberty. J Clin Endocrinol Metab 1998; 83: 2260–2265.
Matkovic V, Ilich JZ, Skugor M, Badenhop NE, Goel P, Clairmont A, Klisovic D, Nahhas RW, Landoll JD . Leptin is inversely related to age at menarche in human females. J Clin Endocrinol Metab 1997; 82: 3239–3245.
Kratzsch J, Lammert A, Bottner A, Seidel B, Mueller G, Thiery J, Hebebrand J, Kiess W . Circulating soluble leptin receptor and free leptin index during childhood, puberty, and adolescence. J Clin Endocrinol Metab 2002; 87: 4587–4594.
Smith CP, Dunger DB, Williams AJ, Taylor AM, Perry LA, Gale EA, Preece MA, Savage MD . Relationship between insulin, insulin-like growth factor I, and dehydroepiandrosterone sulfate concentrations during childhood, puberty and adult life. J Clin Endocrinol Metab 1989; 68: 932–937.
Remer T, Manz F . Role of nutritional status in the regulation of adrenarche. J Clin Endocrinol Metab 1999; 84: 3936–3944.
Genazzani AR, Pintor C, Corda R . Plasma levels of gonadotropins, prolactin, thyroxine and adrenal and gonadal steroids in obese prepubertal girls. J Clin Endocrinol Metab 1978; 47: 974–979.
De Simone M, Farello G, Palumbo M, Gentile T, Ciuffreda M, Olioso P, Cinque M, De Matteis F . Growth charts, growth velocity and bone development in childhood obesity. Int J Obes Relat Metab Disord 1995; 19: 851–857.
Biason-Lauber A, Zachmann M, Schoenle EJ . Effect of leptin on CYP17 enzymatic activities in human adrenal cells: new insight in the onset of adrenarche. Endocrinology 2000; 141: 1446–1454.
Cizza G, Dorn LD, Lotsikas A, Sereika S, Rotenstein D, Chrousos GP . Circulating plasma leptin and IGF-1 levels in girls with premature adrenarche: potential implications of a preliminary study. Horm Metab Res 2001; 33: 138–143.
l'Allemand D, Schmidt S, Rousson V, Brabant G, Gasser T, Gruters A . Association between body mass, leptin, IGF-1 and circulating adrenal androgens in children with obesity and premature adrenarche. Eur J Endocrinol 2002; 146: 537–543.
Schmidt H, Schwarz HP . Premature adrenarche, increased growth velocity and accelerated bone age in male patients with Prader–Labhart–Willi syndrome. Eur J Pediatr 2001; 160: 69–70.
Eiholzer U, Stutz K, Weinmann C, Torresani T, Molinari L, Prader A . Low insulin, IGF-1 and IGFBP-3 levels in children with Prader–Labhart–Willi syndrome. Eur J Pediatr 1998; 157: 890–893.
I'Allemand D, Eiholzer U, Rousson V, Girard J, Blum W, Torresani T, Gasser T . Increased adrenal androgen levels in patients with Prader–Willi syndrome are associated with insulin, IGF-I, and leptin, but not with measures of obesity. Horm Res 2002; 58: 215–222.
Sorva R . Children with craniopharyngioma: early growth failure and rapid postoperative weight gain. Acta Pediatr Scand 1988; 77: 587–592.
Geffner ME . The growth without growth hormone syndrome. Endocrinol Metab Clin North Am 1996; 25: 649–663.
Vignolo M, Naselli A, Di Battista E, Mostert M, Aicardi G . Growth and development in simple obesity. Eur J Pediatr 1988; 147: 242–244.
Vanderschueren-Lodeweyckx M . The effect of simple obesity on growth and growth hormone. Horm Res 1993; 40: 23–30.
Veldhuis JD, Iranmanesh A, Ho KKY, Waters MJ, Jhonson ML, Lizarradle G . Dual effect in pulsatile growth hormone secretion and clearance subserve the hyposomatotropism of obesity in man. J Clin Endocrinol Metab 1991; 72: 51–59.
Scacchi M, Pincelli AI, Cavagnini F . Growth hormone in obesity. Int J Obes Relat Metab Disord 1999; 23: 260–271.
Kopelman PG, Noonan K, Goulton R, Forrest AJ . Impaired growth hormone response to growth hormone releasing factor and insulin-hypoglycemia in obesity. Clin Endocrinol (Oxf) 1985; 23: 87–94.
Camanni F, Ghigo E . Relationships between IGF-1 and age, gender, body mass, fat distribution, metabolic and hormonal variables in obese patients. Int J Obes Relat Metab Disord 1999; 23: 612–618.
Park MJ, Kim HS, Kang JH, Kim DH, Chung CY . Serum levels of insulin-like growth factor (IGF-1), free IGF-1, IGF-binding protein (IGFBP-1), and IGFBP-3 and insulin in obese children. J Pediatr Endocrinol Metab 1999; 12: 139–144.
Attia N, Tamborlane WV, Heptulla R, Maggs D, Grozman A, Sherwin RS, Caprio S . The metabolic syndrome and insulin-like growth factor I regulation in adolescent obesity. J Clin Endocrinol Metab 1998; 83: 1467–1471.
Bucher H, Zapf J, Torresani T, Prader A, Froesch ER, Illig R . Insulin-like growth factors I and II, prolactin and insulin in 19 growth hormone-deficient children with excessive, normal or decreased longitudinal growth after operation for craniopharyngioma. N Engl J Med 1983; 309: 1142–1146.
Bistritzer T, Lovchik JC, Chalew SA, Kowarski AA . Growth without growth hormone: the ‘invisible’ GH syndrome. Lancet 1988; 1: 321–323.
Maor G, Rochwerger M, Segev Y, Phillip M . Leptin acts as a growth factor on the chondrocytes of skeletal growth centers. J Bone Mineral Res 2002; 17: 1034–1043.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shalitin, S., Phillip, M. Role of obesity and leptin in the pubertal process and pubertal growth—a review. Int J Obes 27, 869–874 (2003). https://doi.org/10.1038/sj.ijo.0802328
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ijo.0802328
Keywords
This article is cited by
-
Trend in age at menarche and its association with body weight, body mass index and non-communicable disease prevalence in Indonesia: evidence from the Indonesian Family Life Survey (IFLS)
BMC Public Health (2022)
-
The role of leptin and low testosterone in obesity
International Journal of Impotence Research (2022)
-
Serum IGF1 and linear growth in children with congenital leptin deficiency before and after leptin substitution
International Journal of Obesity (2021)
-
Clinical, laboratory and radiological assessment of skeletal maturation in children and adolescents with obesity
Egyptian Pediatric Association Gazette (2020)
-
Emotion regulation training in the treatment of obesity in young adolescents: protocol for a randomized controlled trial
Trials (2020)
