

Key Points
- Lower esophageal sphincter (LES) circular smooth muscle utilizes two distinct intracellular pathways: (1) a protein kinase C (PKC)-dependent pathway activated by low doses of agonists or during maintenance of spontaneous tone, and (2) a Ca2+-calmodulin-myosin light chain kinase (MLCK)-dependent pathway activated in response to maximally effective doses of agonists during the initial phase of contraction.
- The Ca2+ levels, released by agonist-induced activity of phospholipase C, determine which contractile pathway is activated.
- The PKC-dependent signaling pathway is activated by prostaglandin F2
 (PGF2) and by thromboxanes during sustained LES contraction. This response may involve activation of the monomeric G-protein RhoA.
(PGF2) and by thromboxanes during sustained LES contraction. This response may involve activation of the monomeric G-protein RhoA.
- Ca2+-calmodulin-MLCK-dependent pathway is activated by PGF2, by thromboxane analogues, and by high doses of agonists, such as acetylcholine (ACh), during the initial contractile phase.
- In LES circular muscle a low molecular weight pancreatic-like phospholipase A2 (group I PLA2) causes production of arachidonic acid, which is metabolized to prostaglandins and thromboxanes.
- In esophagitis, inflammation of the mucosa causes production of specific inflammatory mediators [e.g., interleukin-6 (IL-6) and platelet-activating factor (PAF)] that diffuse to the circular muscle and produce mediators such as H2O2. H2O2 diffuses across biologic membranes and leads to production of additional inflammatory mediators such as IL-1
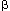 .
.
- IL-1 and other inflammatory mediators may enhance further production of H2O2 and also affect LES signal transduction mechanisms, leading to suppression of sustained contraction.
- In esophagitis, the releasable Ca2+ stores are depleted and that may impair activation of the Ca2+-dependent PKC. Lipid peroxidation may result in production of 8-iso-PGF2 that antagonizes the endogenously produced PGF2. Moreover, smooth muscle relaxants such as PAF and PGE2
 are produced in increased quantities.
are produced in increased quantities.
Signal Transduction in Normal Lower Esophageal Sphincter (LES) Circular Muscle
The body of the esophagus is normally relaxed and contracts only briefly when required to produce peristalsis. In contrast, the lower esophageal sphincter (LES) maintains a sustained pressure, and relaxes to allow the passage of a bolus. The neuromuscular mechanisms that participate in the physiologic regulation of these functions are not well understood. It is thought that LES tone is spontaneous and regulated mostly through myogenic mechanisms, whereas LES relaxation and esophageal contraction are induced by neural mechanisms. In recent years, considerable attention has been given to neural mechanisms regulating gastrointestinal motor function, but relatively little information is available on the intracellular signal transduction pathways responsible for contraction of either esophagus or LES smooth muscle. This review discusses the mechanisms responsible for initiating contraction of LES, the maintenance of LES tone, and how these mechanisms are altered in animals or patients with esophagitis.
Differences in the signal transduction pathways mediating the initial and the sustained phases of contraction have recently been demonstrated1, 2, 3 in gastrointestinal smooth muscle. We find that intracellular pathways utilized by LES muscle to initiate contraction in response to high doses of agonists are different from those utilized in response to low doses of agonists or to maintain tone in sustained contraction. These differences are particularly relevant to LES function, where both initial and sustained contraction may occur as part of normal LES function.
Our data suggest the existence of two distinct contractile signal-transduction pathways in LES smooth muscle cells. Activation of either one may be regulated by the concentration of intracellular Ca2+. The level of intracellular Ca+ available for contraction determines which of the two pathways will be activated. A switch from one pathway to the other can occur3, 4, 5, 6 if releasable Ca2+ levels change.
LES Contraction
MLCK-Dependent Contractile Pathways in LES Circular Smooth Muscle
One of the two pathways, for instance the one activated by maximal doses of acetylcholine (ACh), activates M3 muscarinic receptors linked to Gq/11–type G proteins to stimulate phosphatidylinositol-specific phospholipase C (PI-PLC), which results in the formation of equimolar concentrations of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 causes the release of significant amount of Ca2+ from intracellular stores. The high level of intracellular Ca2+ then promotes the formation of calcium-calmodulin complex, leading to myosin light chain phosphorylation and muscle contraction, as shown in Figure 1. Calmodulin, in the presence of elevated calcium concentrations, inhibits PKC and activates myosin light chain (MLC) kinase (MLCK), resulting in phosphorylation of myosin light chains and contraction.7 This protein kinase C (PKC)-independent, MLCK-dependent pathway has been well described for LES in response to ACh and other agonists8, 9 and for other smooth muscles.10, 11
Figure 1: Signal transduction for lower esophageal sphincter (LES) contraction in response to agonists.
Contraction of LES cells by maximally effective dose of acetylcholine (ACh) is shown as an example of signal transduction in response to high doses of agonists. ACh-induced contraction is mediated by activation of M3 muscarinic receptors linked to Gq and to phosphatidylinositol-specific phospholipase C (PI-PLC), and production of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol bisphosphate (PIP2). IP3 causes release of Ca2+ from stores (ER=endoplasmic reticulum) at a concentration sufficient to cause activation of calmodulin (CAM). Ca2+-CAM activates myosin light chain kinase (MLC Kinase) and inhibits protein kinase C (PKC), inducing a contraction that is entirely CAM dependent. Ca2+-CAM–induced inhibition of PKC masks the presence of other factors that would otherwise contribute to activation of PKC. (Source: Harnett et al.,3 with permission from the American Physiological Society.)
PKC-Dependent Contractile Pathways in LES Circular Smooth Muscle
A second pathway involves a PKC-dependent mechanism of contraction and occurs with submaximal levels of agonists, or in maintenance of tone. Low levels of agonists cause submaximal activation of phospholipases. Activation of specific phospholipases may depend on the agonist, their receptors, and the associated G proteins. For instance, low-level ACh causes submaximal PI-PLC activity, producing low levels of IP3, which releases low levels of Ca2+ from intracellular stores. The mildly elevated intracellular Ca2+ levels are insufficient to activate calmodulin, which requires almost micromolar Ca2+ concentrations4 and MLCK, which is critically dependent on activation of calmodulin.
However, nanomolar Ca2+ levels are sufficient to interact with DAG, generated by PI-PLC and by phosphatidylcholine-specific PLC (PC-PLC) that is concurrently activated by submaximal ACh, to produce submaximal contraction through a PKC-dependent pathway. A PKC-dependent, MLCK-independent pathway may be activated during sustained (or tonic) LES contraction, which is also PKC dependent.12
In the tonically contracted LES, activation of this PKC-dependent pathway, which involves the Ca2+-sensitive PKC,13 may be initiated by activation of at least one (a pancreatic-like or group I) secreted phospholipase A2 (sPLA2),14, 15 resulting in production of arachidonic acid (AA) (Figure 2) and arachidonic acid metabolites, such as PGF2 and thromboxane A2 (TXA2).
Figure 2: Signal transduction for LES tone.

LES tone depends on activity of a group I secreted phospholipase A2 (sPLA2) producing arachidonic acid (AA), which is metabolized to prostaglandin F2 (PGF2) and thromboxane A2 (TXA2). PGF2 and TXA2 may cross the cytoplasm and activate membrane receptors linked to Gq and Gi3. Gq activates phosphatidylinositol-specific phospholipase C (PI-PLC) to produce diacylglycerol (DAG) and IP3-induced Ca2+ release. Gi3 activates phosphatidylcholine-specific phospholipase C (PC-PLC) to produce DAG. Ca2+ and DAG activate PKC resulting in phosphorylation of myosin light chain and contraction. (Source: Harnett et al.,3 with permission from the American Physiological Society.)
Activity of sPLA2, resulting in generation of AA, PGF2, and TXA2, may be the initiating event responsible for sustained maintenance of LES tone. If this hypothesis is correct, activity of sPLA2 results in formation of AA, which is membrane permeable and diffuses across the cytoplasm to be metabolized to PGF2 and TXA2 or TXB2, by appropriate enzymes inside the LES muscle cells. PGF2 and TXA2/B2 may then interact with specific G-protein–linked cell receptors, to activate PI-PLC and PC-PLC, resulting in formation of more DAG than IP3. The relatively modest IP3-induced Ca2+ release may facilitate activation of the Ca2+-dependent PKC by the substantially higher concentrations of DAG produced by the combined activities of PI-PLC and PC-PLC.
In the following subsections we discuss in some detail some of the steps leading to sustained contraction of LES circular muscle.
sPLA2-Induced Formation of AA
Figure 3 shows that, when LES circular muscle is incubated in Krebs solution containing [3H]AA to label AA-containing phospholipids, then washed to remove unincorporated AA, LES circular muscle has a higher AA uptake and release than esophageal circular muscle, which does not maintain tone. The higher AA production is most likely due to activity of a noncytosolic PLA2 because release of AA and LES resting tone are reduced by a selective inhibitor of group I (pancreatic) PLA216 and not affected by the selective cytosolic PLA2 inhibitor AACOCF3,15 as shown in Figure 4.
Figure 3: [3H]Arachidonic acid (AA) content and release of LES and esophageal circular smooth muscle.
![Figure 3 : [3H]Arachidonic acid (AA) content and release of LES and esophageal circular smooth muscle. Unfortunately we are unable to provide accessible alternative text for this. If you require assistance to access this image, or to obtain a text description, please contact npg@nature.com](/gimo/contents/pt1/thumbs/gimo24-f3.jpg)
LES circular muscle tissue was incubated for 4 hours in Krebs solution containing [3H] arachidonic acid (3  Ci/mL), to allow uptake into the muscle, then rinsed to remove unincorporated [3H]AA. After 60 minutes, the radioactivity released into the medium (b) was measured, and compared to the radioactivity present in tissue homogenates (a). Arachidonic acid levels were significantly higher in LES circular muscle than in esophageal circular muscle, which does not maintain tone, indicating a higher uptake of AA in LES muscle. LES circular muscle released significantly higher concentrations of AA than the esophagus, indicating a higher rate of metabolism of AA-containing phospholipids by the LES. Values are mean
Ci/mL), to allow uptake into the muscle, then rinsed to remove unincorporated [3H]AA. After 60 minutes, the radioactivity released into the medium (b) was measured, and compared to the radioactivity present in tissue homogenates (a). Arachidonic acid levels were significantly higher in LES circular muscle than in esophageal circular muscle, which does not maintain tone, indicating a higher uptake of AA in LES muscle. LES circular muscle released significantly higher concentrations of AA than the esophagus, indicating a higher rate of metabolism of AA-containing phospholipids by the LES. Values are mean  standard error (SE) of five to seven animals.
standard error (SE) of five to seven animals.
Figure 4: Secreted phospholipase A2 (sPLA2) inhibitors and [3H]AA release and LES tone.
![Figure 4 : Secreted phospholipase A2 (sPLA2) inhibitors and [3H]AA release and LES tone. Unfortunately we are unable to provide accessible alternative text for this. If you require assistance to access this image, or to obtain a text description, please contact npg@nature.com](/gimo/contents/pt1/thumbs/gimo24-f4.jpg)
a: LES muscle was incubated with [3H] arachidonic acid (3 Ci/mL), and then washed to remove unincorporated [3H]AA, and incubated in Krebs alone (control), AM5 or MJ33 (10-4 M). [3H]AA release, an index of spontaneous PLA2 activity, was significantly reduced by the nonselective sPLA2 inhibitor AM5, and by the selective group I PLA2 inhibitor MJ33. B: In LES muscle strips the group I PLA2 inhibitor MJ33 decreased LES tone, and tetrodotoxin (10-4 M) had no effect on the decrease. The cytosolic PLA2 inhibitor AACOCF3 had no effect excluding involvement of cytosolic PLA2. These data suggest that group I sPLA2 may play a role in the maintenance of LES tone and that this PLA2 may not come from nerve terminals. Values are means + SE of three to six animals.
Group 1 or pancreatic sPLA2, was originally identified in pancreatic juice and then identified and cloned in tissues, including spleen, lung, ovary, and kidney.17, 18, 19, 20, 21, 22 Other secreted PLA2 have been recently discovered. The sPLA2 family comprises 14 to 16 kd, calcium-dependent interfacial enzymes that liberate sn-2 fatty acids including AA for the biosynthesis of eicosanoids.19, 23, 24, 25 Ten sPLA2 enzymes have been described in mammals that currently include group IB, IIA, IIC, IID, IIE, IIF, III, V, X, and XII sPLA2.26, 27 These sPLA2 have several features distinct from other PLA2 families, such as a high disulfide bond content, a requirement for millimolar concentration of Ca2+ for catalysis, and a broad specificity for phospholipids with different polar head groups and fatty acyl chains.26, 28 These enzymes exert biologic effects through multiple mechanisms, including release of AA, which may be metabolized to leukotrienes and prostaglandins,29 bactericidal activity via hydrolysis of the outer membrane of gram-positive bacteria,30, 31and binding to specific sPLA2 receptors.32, 33, 34 Groups I, II, V, and X PLA2s are structurally related, and have a highly conserved Ca2+-binding loop (XCGXGG) and catalytic site (DXCCXXHD).
We used degenerate primers, taken from mostly conserved regions of the published pancreatic PLA2 sequences of human, pig, bovine, mouse, dog, and rat to amplify by reverse-transcriptase polymerase chain reaction (RT-PCR) and then sequence cat pancreatic PLA2. The resulting messenger RNA (mRNA) of cat pancreatic sPLA2 had 555 bases and 89% identity with dog, 84% with human and pig, 81% with rat, and 80% with mouse. Primers for human and cat pancreatic PLA2s were then used to obtain human and cat LES PLA2s, respectively. A group 1B PLA2 was found in the circular smooth muscle layer of cat and human LES and exhibited complete mRNA identity with pancreatic PLA2. Group I PLA is present in the cat internal anal sphincter, the pyloric sphincter, and the colon, suggesting that sPLA2 may be present in sphincters and other gastrointestinal smooth muscles.35
Similarly we cloned by RT-PCR groups IIA and V sPLA2 from the human LES circular muscle layer. The presence of the related group X PLA2 has not yet been tested. The cellular localization of these enzymes remains to be established and their relative importance for production of AA in maintenance of tone remains to be determined.
The presence of multiple PLA2 in the same tissue has been reported in duodenum, jejunum, and lung,36 and may participate in AA production in LES circular muscle. Whether the group IIA, V, and X enzymes have a physiologic contractile function in LES muscle remains to be demonstrated.
AA Metabolites (PGF2a, TXA2/B2) and LES Tone
The AA produced by sPLA2 in the LES is metabolized to prostaglandins, such as PGF2 and thromboxanes, which, in turn contribute to maintaining tone because the cyclooxygenase inhibitors indomethacin [but not the lipoxygenase inhibitor nordihydroguaiaretic acid (NDGA)] reduced basal LES tone (Figure 5). The inhibition was myogenic, as it was not affected by the neural blocker TTX.15
Figure 5: Cyclooxygenase or lipoxygenase inhibitors and LES basal tone.

LES circular muscle strips were incubated in the lipoxygenase inhibitor nordihydroguaiaretic acid (NDGA) or the cyclooxygenase inhibitor indomethacin (Indo), alone or in the presence of tetrodotoxin (TTX) 10-4 M. NDGA had no effect. Indomethacin caused a concentration-dependent reduction in LES basal tone that was not affected by tetrodotoxin. These data suggest that prostaglandins, and not leukotrienes, play a role in LES basal tone and that prostaglandins may not come from nerve terminals. Values are mean SE of three to seven animals. (Source: Cao et al.,15 with permission from the American Physiological Society.)
The PGF2 content of unstimulated LES circular muscle is significantly higher than in esophageal muscle, which does not maintain tone. Indomethacin significantly reduces both tone and PGF2 formation in LES smooth muscle, suggesting that PGF2 may participate in the maintenance of LES resting tone. Thromboxane A2 cannot be tested directly because it is an unstable arachidonate metabolite, with a half-life of 30 seconds, and rapidly decays nonenzymatically to the stable TXB2, which has weak biologic activity,37 but the thromboxane analogue U46619 and the stable TXB2 dose-dependently contracts LES strips and the TXA2 antagonist SQ-29548 decreases resting LES tone, suggesting that TXA2 and/or TXB2 may also contribute to maintaining LES tone.15
The action of these prostanoids is mediated by distinct G-protein–linked receptors. We have shown that in the LES some G proteins are active, that is, bound to guanosine triphosphate (GTP) in the absence of exogenous stimuli. The same G proteins that are spontaneously active are stimulated by PGF2 and TXB2. In addition, LES basal tone can be concentration dependently reduced by the Gi inhibitor pertussis toxin (PTX), suggesting that basal Gi activation may contribute to LES basal tone. The activity of these G proteins is reduced by indomethacin, supporting the view that spontaneous activation of these G proteins is maintained by cyclooxygenase-catalyzed AA products. Activation of these G proteins may play a role in LES basal tone because concentrations of indomethacin that reduce tone also reduce the level of activity of Gq and Gi3 present in the unstimulated LES circular muscle.
These data, initially obtained in the cat, have been confirmed for human LES circular muscle, from organ donors.14 They support the hypothesis, illustrated in Figure 2, that spontaneous activation of a group I sPLA2 causes production of AA and AA metabolites such as PGF2 and thromboxanes, which maintain activation of G proteins such as Gi3 and Gq. These G proteins are linked to phospholipases such as PI-PLC and PC-PLC, which, in combination, produce IP3 and excess DAG to synergistically activate PKC.13, 38
In muscle cells from the small intestine Murthy et al.2, 39 have proposed that different signal transduction pathways are activated during the initial phase of contraction, as opposed to the following sustained phase of contraction. Thus initial contraction induced by Ca2+, agonists, and growth factors is mediated by MLCK, whereas sustained contraction is mediated by activation of Gq/11 and G13, linked to activation of specific Ca2+-dependent and -independent PKC isozymes. This pathway involves activation of Rho-kinase, and adenosine diphosphate (ADP) ribosylation factor (ARF) and is shown in gray in Figure 6. In this pathway agonist binding to receptors activates G13, inducing sequential activation of a Rho-specific, guanine nucleotide exchange factor (Rho-GEF), RhoA, and then Rho kinase and phospholipase D (PLD). In this pathway PLD produces phosphatidic acid (PA), which is dephosphorylated to DAG40; DAG in turn activates PKC, which causes contraction and may also enhance DAG production by further potentiating PLD activation. Phospholipase D is also activated by an ARF-dependent pathway, but the mechanism of activation of ARF is unknown.9, 39
Figure 6: Prostaglandin F2 (PGF2)-induced protein kinase C (PKC) activation in LES tone and sustained contraction.
(PGF2)-induced protein kinase C (PKC) activation in LES tone and sustained contraction.
![Figure 6 : Prostaglandin F2|[alpha]| (PGF2|[alpha]|)-induced protein kinase C (PKC) activation in LES tone and sustained contraction. Unfortunately we are unable to provide accessible alternative text for this. If you require assistance to access this image, or to obtain a text description, please contact npg@nature.com](/gimo/contents/pt1/thumbs/gimo24-f6.jpg)
We have shown (data in brown and green) that maintenance of LES tone is PKC-dependent, and mediated in part by Gq linked to phosphatidylinositol-specific phospholipase C (PI-PLC) and by Gi3 linked to phosphatidylcholine-specific phospholipase (PC-PLC). The interrupted gray arrows represent a proposed contractile pathway for maintenance of tone and sustained contraction.2 In sustained contraction receptors may activate G13, and initiate a cascade involving activation of a guanine nucleotide exchange factor (Rho-GEF), RhoA, Rho kinase, and phospholipase D (PLD). PLD produces diacylglycerol (DAG), which activates PKC, causing contraction and may potentiate PLD activation. PLD is also activated by an adenosine diphosphate (ADP) ribosylation factor (ARF)-dependent pathway. The mechanisms of activation of Rho kinase–induced activation of PLD and of ARF activation are unknown. (Source: Harnett et al.,3 with permission from the American Physiological Society.)
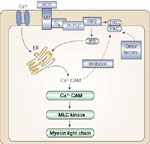
We have confirmed that, in human and cat LES circular muscle strips, a Rho kinase–dependent pathway participates in maintenance of tone, because the Rho kinase inhibitor Y27632 dose-dependently decreases tone, abolishing tone at 10-4M. Exogenously administered PGF2 (or the stable TXA2 analogue U46619) caused sustained contraction in enzymatically isolated cells and in LES muscle strips, lasting more than 30 minutes, which was also reduced by Y27632 (Figure 7). After treatment with the Rho kinase inhibitor, Y27632 cells achieved the same initial shortening, but did not remain contracted, and returned to their unstimulated length within a few minutes. The inhibition of sustained contraction by Y27632 in cells mimics the inhibition of tone or of sustained PGF2- (or TXA2)-induced contraction in strips (Figure 7).
Figure 7: RhoA in LES tone and PGF2-induced sustained contraction.
![Figure 7 : RhoA in LES tone and PGF2|[alpha]|-induced sustained contraction. Unfortunately we are unable to provide accessible alternative text for this. If you require assistance to access this image, or to obtain a text description, please contact npg@nature.com](/gimo/contents/pt1/thumbs/gimo24-f7.jpg)
a: In LES circular muscle cells, PGF2 caused a relatively rapid initial contraction that was maintained in excess of 20 minutes. In the presence of the Rho kinase inhibitor Y27632 the initial phase of contraction in response to PGF2 was not affected. The sustained contraction, however, was almost abolished by Y27632, indicating that the initial and the sustained phase of contraction are mediated through different intracellular pathways. b: The Rho kinase inhibitor Y27632 reduced LES sustained contraction in response to PGF2, confirming the participation of RhoA in maintenance-sustained contraction in muscle strips as well as in isolated muscle cells. (Source: Harnett et al.,3 with permission from the American Physiological Society.)
In addition, preliminary data in LES circular muscle (shown in black in Figure 6) indicate that when cells are permeabilized to allow entrance of antibodies into the cytoplasm, the time course of PGF2- or TXA2-induced shortening is the same as in intact cells. In these permeable cells ARF1 and RhoA antibodies reduce and G13 antibodies abolish sustained contraction induced by PGF2, confirming that the contraction is mediated by RhoA-induced activation of Rho kinase and ARF1.
In previous work, we reported that maintenance of LES tone is PKC dependent,12 that Gq and Gi3 are active15, that Gq is linked to PI-PLC, and Gi3 to PC-PLC,6 and that PI-PLC and PC-PLC are also active12 as tone is maintained. These G proteins are linked to phospholipases such as PI-PLC and PC-PLC, which, in combination, produce IP3 and IP3-induced Ca2+ release, with excess DAG to synergistically activate the Ca2+-sensitive PKC.13, 38 An additional contractile pathway for maintenance of LES tone, and of sustained contraction in response to PGF2, is shown in gray in Figure 6. This pathway is also activated during sustained contraction of intestinal smooth muscle cells in response to CCK39 and during initial contraction of esophageal circular muscle in response to PGF2.9 The data obtained in our laboratory are indicated in black.
All three pathways—PI-PLC-dependent, RhoA-dependent, and PC-PLC dependent—result in activation of PKC.
PKC-Dependent Contraction in Maintenance of LES Tone
The precise mechanisms responsible for mediation of this PKC-dependent contraction are not well established, but there is a clear distinction between calcium-calmodulin-MLCK–dependent contraction and PKC-dependent contraction, which is not dependent on activation of the calcium-sensitive MLCK. The contractile paradigm for the calcium-calmodulin-MLCK–dependent contraction has been understood for some time. Understanding of the mechanisms responsible for the PKC-dependent contractile pathways is still evolving, and considerable confusion in the literature has derived from attempts to explain PKC-dependent contractile pathways as variants of the MLCK-dependent pathway, involving increased sensitization to calcium, inactivation of phosphatases, or other mechanisms.41, 42, 43, 44. In the LES these pathways are clearly distinct and independent of each other: at low calcium levels, which are insufficient to fully activate calmodulin, contraction is PKC-dependent, whereas when calcium levels are sufficient to fully activate calmodulin, the activated calmodulin inhibits PKC and activates MLCK.
We find that the PKC-dependent contractile pathways responsible for maintenance of LES resting tone depend on phosphorylation and activation of the extracellular signal regulated kinases ERK1 and ERK2, and/or a 27-kd heat shock protein (HSP27)-linked p38 kinase.45Figure 8 shows that the ERK1/ERK2 inhibitor PD98059 and the p38MAPK inhibitor SB203580, when used separately, reduce the tone of LES muscle strips, and almost abolish it when used in combination, supporting the view that PKC-dependent contraction of LES circular muscle is almost entirely mediated through activation of these mitogen-activated protein (MAP) kinases.45
Figure 8: Mitogen-activated protein (MAP) kinases mediate PKC-dependent LES basal tone.
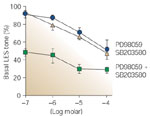
MAPK kinase (MEK) inhibitors prevent phosphorylation and activation of the MAP kinases ERK1 and ERK2. The MEK inhibitor PD98059 or the p38 MAP kinase inhibitor SB203580, when used alone, produced concentration-dependent reductions in LES tone [p = .001, analysis of variance (ANOVA)]. LES tone was further reduced when PD98059 and SB203580 were used in combination (p = .001, ANOVA), suggesting that PKC-dependent tone is mediated by ERK1/2 and p38 MAP kinases. Values are means + SE of three animals. (Source: Cao et al.,45 with permission from the American Physiological Society.)
The MAP kinases comprise a family of serine threonine kinases, which include ERK1 and ERK2, the Jun N terminal kinase/stress activated protein kinase, and p38 MAP kinase.46 In LES circular muscle, ERK1, ERK2, and p38 MAP kinases participate only in the PKC-dependent contractile pathway, and not in the calmodulin-MLCK–dependent pathway.45
These data indicate that PKC-dependent contraction, such as ACh-induced contraction of esophageal circular muscle, or LES spontaneous tone, may be associated with activation of two parallel MAP kinase contractile pathways: an ERK MAP kinase pathway and an HSP27-linked p38 kinase pathway. Protein kinase C may activate the monomeric G protein Raf, causing it to phosphorylate MAPK kinase (MEK), which in turn may phosphorylate Map kinase (MAPK)47; MAPK may then phosphorylate calponin or caldesmon, and other intermediate proteins. We propose that two distinct parallel pathways may contribute to contraction, one involving p38 kinase and HSP27, and the other involving ERKs and calponin/caldesmon.
One of the intermediate proteins in the PKC- MAPK contractile pathway may be integrin-linked kinase (ILK).48 Integrin-linked kinase was identified and cloned based on its interaction with the 1 integrin cytoplasmic domain,49 and its catalytic domain is homologous with other protein kinase catalytic domains. Integrin-linked kinase interacts with various proteins, several of which are connected to the actin cytoskeleton, and anchor it to the extracellular matrix.
Integrin-linked kinase is expressed in most mammalian cells, with high expression levels in cardiac and skeletal muscle, and its sequence is highly conserved across species. Some ILK are linked to integrins localized to membrane-associated dense plaques, which are structurally similar to the focal adhesion sites of cultured cells.50, 51 These membrane sites have long been identified as the sites of tension transmission between the contractile apparatus and the extracellular matrix.52 A second population of ILK, which is Ca2+-independent, is associated with myofilaments and may be responsible for Ca2+-independent (i.e., MLCK-independent) phosphorylation of myosin in smooth muscle preparations.47 The report that purified myosin contains a low level of Ca2+-independent MLC20 kinase activity that is resistant to the MLCK inhibitor AV25 supports the association of ILK with myosin filaments.48 Integrin-linked kinase directly phosphorylates myosin light chain on serine 18 and threonine 19, which are also phosphorylated by MLCK.48
Preliminary data from our laboratory show that ILK is present in esophageal and LES smooth muscle and that an ILK antibody partially inhibits (approximately 50%) ACh-induced contraction of saponin-permeabilized esophageal cells. In addition, preliminary data suggest that ILK participates in the ERK1/ERK2 and not in the p38 MAP kinase contractile pathway. The mechanisms of activation of the plasma membrane integrin-linked ILK have been described to some extent,53, 54 but regulation of myofilament-linked ILK has not yet been elucidated.
Integrin-linked kinase may be the kinase responsible for MLC phosphorylation in the ERK1/ERK2 pathway, as illustrated in Figure 9.
Figure 9: Protein kinase C (PKC)-mediated contraction of LES circular muscle.
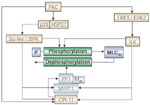
PKC-mediated contraction may depend on activation of at least two pathways: an ERK1/ERK2 MAP kinase pathway linked to the Ca-independent integrin-linked kinase (ILK), and a 27-kd heat shock protein (HSP27)-p38 kinase pathway linked to the Ca-independent zipper interacting protein (ZIP)-like or ZIP-kinase. ILK, ZIP-like, or ZIP kinase may directly phosphorylate myosin light chains (MLC20), and they may phosphorylate the targeting subunit of myosin phosphatase (MYPT1) or CPI-17, inhibiting phosphatase activity. CPI-17 (17-kd PKC-dependent phosphatase inhibitor) may also be directly phosphorylated by PKC. (Source: Harnett et al.,3 with permission from the American Physiological Society.)
The kinase responsible for MLC phosphorylation in the p38-HSP27 pathway has not yet been defined, but a reasonable candidate may be zipper interacting protein (ZIP) kinase (ZIPK), which was isolated from a HeLa cell complementary DNA (cDNA) library, with an amino acid sequence identical to that of a ZIPK. Zipper interacting protein kinase phosphorylates the regulatory light chain of myosin II at both serine 19 and threonine 18 in a Ca2+/calmodulin independent manner,55and thus may be a candidate as an intermediate protein in MLCK-independent contraction. Niiro and Ikebe56 found that ZIPK is expressed in smooth muscle tissues and that it can phosphorylate myosin in a Ca2+-independent manner and thereby induce Ca2+-independent contraction of permeabilized smooth muscle. In a pilot Western blot, using an antibody raised against the C-terminal of ZIPK, in esophageal and LES circular muscle we find a band at 52 kd, which is the appropriate molecular weight and a ZIPK antibody partially inhibits (approximately 50%) ACh-induced contraction of esophageal cells.
In addition, a ZIP-like kinase that is closely related to ZIPK has been identified57 and shown to phosphorylate the targeting subunit of myosin phosphatase (MYPT1). This kinase, which is also referred to as MYPT1 kinase,58 may be a truncated version of ZIPK, or an independent relative. If not identical, the two kinases are closely related as evidenced by cross-reactivity of ZIP-like kinase with some ZIPK antibodies and by similarities in enzyme properties.58 The molecular basis underlying the interaction of ZIP-like kinase with the targeting subunit of MYPT1 is not known, but it is thought that ZIP-like kinase is cytosolic and associated with MYPT1 and myosin.59 This ZIP-like kinase phosphorylates MYPT1 and inhibits phosphatase activity.
Inhibition of phosphatases is a regulatory mechanism of smooth muscle contraction.41, 42, 60, 61 Inhibition of phosphatases in response to appropriate stimuli potentiates myosin phosphorylation, by MLCK or by other kinases in the PKC-dependent pathway, such as ILK, ZIP kinase, and ZIP-like kinase.
Based on these published and unpublished data from our lab, we propose that LES tone depends on sustained contractile mechanisms, which are distinct from those activated in the initial phase of contraction. Lower esophageal sphincter tone depends on sustained production of AA by a group I PLA2 and possibly by other PLA2s. Arachidonic acid, in turn, is metabolized to PGF2 and thromboxanes, which maintain tone by inducing sustained contraction through activation of PGF2 and thromboxane receptors. The mechanism of receptor-induced sustained contraction has not been completely elucidated for LES circular muscle, but is likely to depend on activation of PKC through several pathways, including activation of PI-PLC, PC-PLC, and Rho-A.
Once PKC is activated, the PKC-dependent pathways resulting in sustained contraction remain to be established. It may involve several intermediate proteins capable of either directly phosphorylating MLC20, or of regulating the activity of MLC phosphatases.
Inflammation-Induced Changes in Signal Transduction in LES Circular Muscle
In Vivo Esophageal Inflammation
Gastroesophageal reflux disease (GERD) is by far the most common esophageal disorder. It may lead to the development of serious complications including ulcers, strictures, bleeding, columnar metaplasia (Barrett's esophagus), and eventually adenocarcinoma of the esophagus.
The pathophysiology of GERD has been intensely investigated and its multifactorial nature is well recognized. The current belief is that GERD results from impaired esophageal defenses due to defective motor function responsible for antireflux and luminal clearance. Reflux of gastric contents may occur as a result of transient lower esophageal sphincter relaxation (TLESR)61 unrelated to swallowing or to secondary peristalsis.63, 64, 65 In the early stages of GERD, TLESR accounts for the largest proportion of reflux episodes,63, 65, 66 but TLESR-unrelated reflux increases with progressive severity of the disease. Impairment of LES tone and esophageal contraction become more prevalent and important in the pathogenesis of GERD as the severity of the disease increases.65, 67
Impairment of these motor functions coupled with decreased tissue resistance may increase the likelihood of further reflux episodes and of impaired acid clearance, aggravating their damage. The spiral of damage leading to worse damage may contribute to permanent impairment of LES tone and of esophageal peristalsis.
Little attention, so far, has been given to the process of esophageal inflammation, even though many substances considered critical to reflux esophagitis are classical inflammatory products, such as cytokines, prostanoids, and reactive oxygen species (ROS). These products are thought to derive from inflammatory cells infiltrating acid-damaged tissue68and may act upon muscle cells and cause them to produce their own cytokines,69 perhaps creating a vicious circle that contributes to and maintains the motility disorders found in gut inflammation.70 Examining the relationship between inflammatory mechanisms and mechanisms responsible for LES tone should help in understanding the pathophysiologic events associated with esophageal disease.
An animal model of acute esophagitis has been used for some time in our laboratory, obtained by perfusing the cat esophagus with 0.1 N HCl for 45 minutes during three successive days and with experiments carried out on the fourth day. In this model of acute esophagitis, we have shown that in vivo LES resting pressure and in vitro LES tone are significantly reduced due to the presence in the circular muscle layer of inflammatory mediators [IL-1 IL-6, and PAF, but not tumor necrosis factor- (TNF-) induced by inflammation and cell damage. In animals with esophagitis these inflammatory mediators cause production of high levels of H2O2.
Addition of H2O2 to normal muscle produces a concentration-dependent decrease of in vitro LES tone by several mechanisms. Reactive oxygen species have been shown to consistently depress the Ca2+–adenosine triphosphatase (ATPase) responsible for uptake of Ca2+ into the endoplasmic reticulum71, 72, 73, 74, 75 and they do so in LES circular muscle.76 In addition to inhibiting Ca2+ uptake into the endoplasmic reticulum, ROS cause Ca2+ release from intracellular stores through both ryanodine- and IP3-sensitive Ca2+ channels.77 The combination of decreased Ca2+ uptake and increased Ca2+ release results in depletion of releasable calcium stores, which may play a role in maintenance of tone by potentiating activation of the Ca2+-sensitive PKC. In addition, H2O2 increases production of lipid mediators, such as PGE2 and PAF, which relax LES circular muscle, and of products of lipid peroxidation such as isoprostanes 8-iso F2, which inhibit PGF2-mediated contraction.78
In this feline model, however, LES function returns to normal within 3 weeks after suspension of acid perfusion,79 whereas in humans, even after healing of the mucosa by endoscopic and histologic criteria, LES and esophageal function do not return to normal.67, 80, 81, 82, 83, 84 It is likely that inflammatory mechanisms may produce time-dependent damage and irreversible alterations in motor function. Using an animal model of myotomy-induced experimental esophagitis, we demonstrated that ranitidine added at 6 months after sphincterotomy restored LES muscle function, suggesting that LES muscle function may be reversible in the early stages before permanent damage takes place.85 In this model, onset of chronic esophagitis may occur after more than 6 months post-LES myotomy.85
Over the course of several years, we have obtained some human esophageal specimens, and we have extended results initially obtained in cat to the human LES. We found that, similarly to the cat, AA contributes to maintenance of human LES tone by producing prostaglandins and thromboxanes.13 Human esophageal specimens from organ donors with reflux esophagitis are exceedingly infrequent and rarely available for study. We were lucky to obtain a human LES specimen with histologically proven erosive esophagitis from an organ donor and characterized its pathophysiologic mechanical and inflammatory profiles.86
When mounted in a warm (37°C) oxygenated muscle chamber, normal human LES circular muscle strips gradually develop tone and reach a steady-state tone after 3 to 4 hours. In contrast, circular muscle strips from the esophagitis LES developed remarkably little tone, but when exposed to the H2O2 scavenger catalase (800 U/mL), tone increased within 15 to 30 minutes and reached a value similar to that of normal LES strips (Figure 10a). These findings suggest that H2O2 may be present in esophagitis LES circular muscle, and responsible for reduced tone when inflammation is present, and are consistent with the finding that IL-1 levels are elevated in the circular smooth muscle layer and lead to an increased production of H2O2 in the human esophagitis specimen (Figure 10b) and in the cat model of experimental esophagitis.
Figure 10: H2O2 and LES.
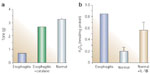
a: Human LES muscle mounted in muscle chambers, near conditions of optimum force development, developed steady tension after 3 to 4 hours (normal). An esophagitis specimen (esophagitis), obtained from an organ donor, had lower tone than normal strips, but tone increased after incubation in catalase.suggesting that H2O2 may be responsible for the reduced tone in esophagitis. b: Treatment of normal LES muscle with IL-1 (200 U/mL, 2 hours) increased H2O2, and H2O2 levels were fourfold higher in LES muscle from the esophagitis specimen compared with normal muscle. Means + SE are shown for three normal LES samples. (Source: Cheng et al.,86 with permission from American Gastroenterological Association.)
Interleukin-1 activates IL-1 receptors, a subfamily of IL-1 Toll-like receptors87 that can result in the activation of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. We have demonstrated that the inflammatory cytokine IL-1 increases production of H2O2 in normal human LES muscle.86 It is possible that in chronic reflux esophagitis elevated levels of cytokines such as IL-1 may cause upregulation of NADPH oxidase, which would maintain elevated levels of H2O2 in the circular smooth muscle layer.
The restoration of tone of the esophagitis specimen by catalase supports the hypothesis that upregulation of nonphagocytic NADPH oxidases present in LES circular muscle is a possible mechanism for overproduction of H2O2.
Phagocytic NADPH oxidase88 consists of six subunits that are partitioned between different subcellular locations in the resting state. Two of these subunits, p22phoxand gp91phox, are integral membrane proteins and form a heterodimeric flavocytochrome, also known as cytochrome b558, which constitutes the catalytic core of the enzyme. The remaining oxidase components reside in the cytosol and include the small guanosine triphosphatase (GTPase) Rac as well as a complex of p40phox, p47phox, and p67phox. Activation of NADPH oxidase is initiated by phosphorylation of phox proteins, which is believed to induce conformational changes that lead to rearrangements, affecting interactions within the cytosolic p40-p47-p67phox complex. These events culminate in the translocation of this complex to the membrane and association with both Rac–guanosine triphosphate (GTP) and cytochrome b558 to form the active enzyme,87 producing superoxide, which is rapidly transformed into the more stable H2O2.
Superoxide-generating homologues of phagocytic gp91phox (NOX1-NOX5, DUOX1, DUOX2) and homologues of other subunits, such as p41 or NOXO1 (homologue of p47phox), p51, or NOXA1 (homologue of p67phox) have been found in several cell types.88, 89, 90 The presence of these homologues in nonphagocytic cells suggests that ROS generated in these cells may have a nonmitochondrial origin and have distinctive cellular functions related to immunity, signal transduction, and modification of the extracellular matrix.88
Human LES smooth muscle contains NOX1,2,3,4 and NOX5, but NOX5 cDNA, as measured by real-time PCR, is the only one that is significantly increased by induction of esophagitis (Figure 11). Upregulation of NADPH oxidases and overproduction of free radicals may cause further upregulation of transcription factors, ultimately resulting in alterations of the cell phenotype.
Figure 11: Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in esophagitis.

LES circular muscle was examined by reverse-transcriptase polymerase chain reaction (RT-PCR) for the presence of NADPH oxidases message. We used published primers for NOX1, NOX2, NOX3, NOX4, and NOX5, and found that these components of the NADPH oxidase enzyme are all present in esophageal circular muscle, but only NOX5 mRNA was increased in the human specimen with esophagitis, as shown on the left. Increased NOX5 cDNA in the esophagitis specimen was confirmed by real-time RT-PCR, when compared to normal human esophageal circular muscle, as shown on the right. Means + SE are shown for three normal (control) and three IL-1–treated LES sample. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (Source: Cheng et al.,86 with permission from American Gastroenterological Association.)
Overproduction of H2O2 in human LES affects maintenance of LES tone, through mechanisms similar to those activated in the cat in response to exogenous H2O2 or after induction of acute esophagitis.78
Similarly to the cat esophagitis model, the human esophagitis LES sample had high levels of PGE2, which is known to relax LES smooth muscle.91 In contrast to PGE2 the levels of PGF2, which contribute to the maintenance of LES tone, does not change after induction of experimental esophagitis in the cat. However, the effectiveness of PGF2 is reduced by the presence of elevated levels of isoprostanes found in cat esophagitis LES circular muscle. Isoprostanes are stable prostaglandin-like compounds that are formed enzymatically92 and nonenzymatically in vivo. F2-isoprostanes may bind with varying affinity to PGF2 receptors93 acting as weak agonists,94 and may bind to specific isoprostane receptors.95 Because isoprostanes are not metabolized, prolonged exposure to isoprostanes may eventually cause desensitization or sequestration of PGF2 receptors.93, 94
In cat and in normal human LES muscle strips, 8-iso- PGF2 does cause little contraction and inhibits PGF2-induced contraction. The LES muscle strips from the esophagitis specimen, which contained elevated levels of isoprostanes, did not contract in response to PGF2 but contracted normally in response to ACh. Elevated levels of F2 isoprostanes in the esophagitis LES may inhibit the endogenously produced PGF2 by binding to PGF2 receptors and perhaps by causing receptor internalization and reducing the number of receptors available for activation by PGF2.78, 86
In addition to the production of isoprostanes, oxidative attack of phosphatidylcholines by ROS yields a series of potent PAF mimetics96, 97, 98 termed " PAF-like agents." The structure of these bioactive lipid products differs slightly from PAF whose sn-2 residue is exclusively derived from acetyl–coenzyme A (CoA). The PAF-like agents, created by oxidation of phospholipids,99 activate PAF receptors in a variety of experimental preparations,96, 97, 98, 99 and their biologic activity is completely blocked by specific PAF receptors antagonists.99 Platelet-activating factor concentration dependently decreases normal human LES tone, H2O2 treatment increased PAF levels in normal LES muscle, and PAF levels were elevated in the human esophagitis specimen.
The most common approach to treatment of GERD is acid-suppression therapy. By decreasing the acidity of gastric contents, the refluxed material is rendered less irritating and the symptoms and mucosal injury improve.100 Although mucosal inflammation is decreased, motor dysfunction of the body of the esophagus and LES continues, consistently with an increased and persistent production of H2O2. H2O2 is likely to play a central role in mediating the decrease in LES tone characteristically found in GERD. We demonstrate that scavenging H2O2 reverses this decrease, pointing to inhibition of ROS as a novel therapeutic approach focused on restoring LES function.
An In Vitro Model of Esophageal Inflammation
In the model of in vivo–induced acute esophagitis we have found that the inflammatory cytokines IL-1 and IL-6 (but not TNF-)101 are elevated in the circular muscle layer. In addition, we have examined human endoscopic mucosal biopsies, obtained from the upper, middle, and lower third of the esophagus of patients with and without endoscopic evidence of esophagitis. These biopsies were organ-cultured overnight in a transwell system and the undernatant recovered for cytokine content. The biopsies showed increased concentration of the proinflammatory cytokine IL-6 in the inflamed tissue compared to noninflamed portions of the esophagus or to controls. Interleukin-1 was also elevated when esophageal inflammation was severe.102
The findings in biopsies from patients with reflux esophagitis are consistent with the data obtained in the cat model of in vivo–induced esophagitis and indicate that both acid-induced acute inflammation of normal mucosa and chronic inflammation of mucosa of patients with reflux esophagitis are characterized by the overproduction of the proinflammatory cytokines IL-1 and IL-6. These cytokines, initially produce in the mucosa, may affect circular smooth muscle function.101 They are thought to derive from inflammatory cells infiltrating acid-damaged tissue68 and may produce additional inflammatory mediators that may amplify and perpetuate tissue injury103, 104 by acting on muscle cells and causing them to produce their own inflammatory mediators.69 For these reasons, examining tissues with well-developed inflammation may provide few clues toward understanding how inflammation develops. Thus, defining the sequential production of inflammatory mediators in reflux esophagitis and their tissues of origin seems essential to better understand the genesis of GERD pathophysiology.
We have recently tested an in vitro model of esophageal inflammation.105 In this in vitro model the mucosa/submucosa is freed by removing the circular and longitudinal muscle, and by sharp dissection at the level of the submucosa, and segments of the mucosal tube are tied at both ends, forming a closed sac (Figure 12). The mucosa tube (or sac) has the squamous epithelium on the inside and the submucosa on the outside. The preparation is kept immersed in warm (37°C) oxygenated Krebs solution, at pH 7.4. The sac may be filled with control Krebs solution, or with HCl (pH 5.8). The mucosal sac, when filled with HCl solution, produces and releases inflammatory mediators into the supernatant. The supernatant surrounding the tube may then be applied to circular muscle strips, or used for measurement of inflammatory mediators. The data obtained from a sac of normal esophageal mucosa filled with Krebs or HCl may be compared to a similar sac of esophageal mucosa from animals with the well-established model of experimental esophagitis, induced by in vivo HCl esophageal perfusion over three successive days. The inflammatory mediators produced or released by mucosa from animals with in-vivo–induced esophagitis may be compared to those of mucosal sac preparation after 2 to 3 hours of exposure to intraluminal HCl.
Figure 12: An in vitro model of esophagitis.

The mucosa/submucosa is freed from esophageal muscle, by sharp dissection at the level of the submucosa. Segments of the mucosal tube are tied at both ends, forming a closed sac with the squamous epithelium on the inside and the submucosa on the outside. The sac with HCl (pH 5.8) or Krebs buffer inside, is kept in oxygenated Krebs buffer supernatant for 3 hours (1 mL Krebs buffer/100 mg mucosa). In response to HCl, the mucosal sac produces and releases inflammatory mediators into the supernatant, which may then be applied to muscle strips, or used for measurement of inflammatory mediators. (Source: Cheng et al.,105 with permission from the American Physiological Society.)
This in vitro mucosal sac preparation is designed to distinguish the inflammatory mediators released by the mucosal layer, from those produced in the circular muscle layer in response to substances released by the mucosa. Because the mucosal sac was created by removing the muscle at the level of the submucosa, it is reasonable to assume that anything that is secreted by the sac and collected in the supernatant would have diffused to the circular muscle in an intact esophagus.
We were somewhat surprised to find that the preliminary data obtained with this in vitro model, in which mucosa lacks blood perfusion, are consistent with those obtained from specimens of in-vivo–induced esophagitis. Inflammatory mediators produced by the isolated mucosal sac after 2 to 3 hours of acid exposure are the same as those found in esophageal mucosa and circular muscle of animals with in-vivo–induced esophagitis. Although this in vitro model may have limits, as it does not involve inflammation-induced recruitment of blood-borne immune cells, the model has the advantage of allowing us to distinguish the contributions of mucosa and muscle to the development of inflammation. The model uses mucosa and muscle from normal animals and does not require repeated perfusion over 3 days. Thus it may be appropriate to examine sequential activation of inflammatory events in the mucosa and in the circular muscle layer of esophageal specimens from cat and from human organ donors that are sometimes available. This is an important feature of the model, because human esophagitis specimens are rare.
A difference between the in vitro model and in vivo induction of esophagitis was found in the pH of the in vivo perfusate and that of the solution filling the esophageal sac. The in vivo esophagus was perfused with 0.1 N HCl, whereas the mucosal sac was filled with HCl at pH 5.8, because a pH dose response for the in vitro preparation indicated that maximum formation of IL-1 and IL-6 in the mucosa occurred when pH was kept between 5.8 and 4.8. Production of IL-1 and IL-6 in the mucosa declined when pH was lowered to 4, due to cell death after 3 hours at this pH, as shown by trypan blue exclusion and lactate dehydrogenase release. It is possible that the in vitro mucosa may be affected by relatively small decreases in pH, as it is not perfused by blood and thus forced to the pH of the surrounding medium, without any way of buffering it. In contrast, the in vivo mucosa is continuously perfused and buffered by blood flow, and exposed to saliva at high pH, and thus more capable of resisting larger excursions in luminal pH.
We find that essentially the same inflammatory processes are present in the body of the esophagus and in the LES. This is not surprising, as the inflammation begins with damage to the epithelial cells in the mucosa. The results of the inflammation, however, are different, arising from differences in the contractile signal transduction pathways of LES and esophageal circular muscle.
When esophageal or LES circular muscle is directly exposed to HCl (pH 5.8, 2 to 3 hours), little change in tone or electric field stimulation (EFS)-induced contraction occurs. When the mucosal sac is filled with HCl at the same pH, however, IL-1 and IL-6 and PAF are formed in the mucosa. Initially IL-1 is not released in the supernatant and remains in the mucosa, but IL-6 and PAF are released into the surrounding environment, presumably to affect circular muscle function. Formation and release of IL-6 from the mucosa is secondary to formation and release of PAF (unpublished observations). Release of PAF and IL-6 from the mucosa induces production of H2O2 in the circular muscle, but not in the mucosa (Figure 13) because no H2O2 is found in supernatant of esophageal mucosa (Figure 14), where it would diffuse if it were produced by the mucosal layer.
Figure 13: H2O2 in LES epithelial and circular muscle cells.

H2O2 levels were examined by confocal microscopy using dihydrorhodamine (DHR 123) as a probe for measurement of intracellular H2O2. Left panel: In untreated epithelial cells (control) H2O2 levels were low and did not change in response to HCl or IL-6. An H2O2-treated epithelial cell is shown as a positive control. H2O2 is membrane permeable, and in the H2O2-treated cell, it penetrates the cytoplasm, demonstrating the adequacy of the technique in detecting cytoplasmic H2O2. Right panel: In normal LES smooth muscle cells (control) H2O2 levels were low and were not affected by HCl. Treatment with supernatant of HCl-filled mucosal sac (mucosa-HCl supernatant), or with supernatant of mucosa from esophagitis animals (esophagitis mucosa supernatant), increased H2O2 in muscle cells. Similarly, treatment with IL-6 or platelet-activating factor (PAF) caused an increase in cytoplasmic H2O2 (Source: Cheng et al.,105 with permission from the American Physiological Society.)
Figure 14: Release of H2O2 in mucosa supernatant.
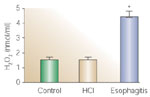
After 3-hour exposure to HCl (pH 5.8) (brown bar) the mucosal sac did not release any more H2O2 into the surrounding supernatant than did nonacidified mucosa (green bar). In contrast, mucosa from animals with in-vivo–induced esophagitis (blue bar) released a significantly higher concentration of H2O2 into the supernatant. Values are mean + SEM, for three animals. (Source: Cheng et al.,105 with permission from the American Physiological Society.)
Exposure to PAF induces upregulation of the NADPH component NOX5 in the circular muscle layer that also appears to be upregulated in the in-vivo–induced esophagitis model. Upregulation of NADPH oxidases and overproduction of free radicals may begin in the circular muscle, but H2O2 may diffuse across biologic membranes106 because the molecule is not charged. Thus H2O2 may diffuse to the mucosal layer as well as through the circular muscle layer. In both tissues H2O2 may cause upregulation of transcription factors and production of additional inflammatory mediators. In the circular muscle layer the presence of H2O2, over time may induce formation of IL-1? explaining the presence of this cytokine in muscle from animals with in vivo esophagitis, induced by 3 days of repeated acid perfusion. In the mucosa layer, back-diffusion of H2O2 from the muscle layer may induce production of H2O2 by the mucosa (Figure 14), explaining the elevated levels of H2O present in the mucosa after induction of in vivo esophagitis by 3 days of acid perfusion.
These data suggest that in vitro contact of HCl with esophageal mucosa stimulates production of IL-6 (in cat) and PAF (in cat and humans), which are released by the mucosa to induce formation of H2O2 by the muscle. H2O2, in turn, may affect esophageal neurons and LES muscle as discussed in the in-vivo–induced esophagitis model. Taken together, the data suggest that development of inflammation may depend on interaction of muscle and mucosa, with both producing and releasing distinct inflammatory mediators.
Conclusion
We find that esophagitis begins in the mucosa, causing production of specific inflammatory mediators (e.g., IL-6 and PAF) that diffuse to the circular muscle. The most significant contribution of the muscle layer in response to these products is H2O2. H2O2 is not charged and free to diffuse across biologic membranes, in the muscle and mucosal layers affecting transcription factors and production of additional inflammatory mediators, including other cytokines such as IL-1
These inflammatory mediators, produced in response to H2O2, by themselves may cause further production of H2O2 and affect LES signal transduction, and its ability to maintain sustained contraction through several mechanisms.
Esophagitis is characterized by overproduction of H2O2 that affects maintenance of LES tone, through several mechanisms: (1) depletion of releasable Ca2+ stores may impair activation of the Ca2+-dependent PKC; (2) H2O2-induced lipid peroxidation results in production of 8-iso-PGF2 and antagonism of the endogenously produced PGF2, which plays a role in maintenance of resting; and (3) H2O2-induced production of other smooth muscle relaxants such as PAF and PGE2 will contribute to decreased LES tone.
