

Key Points
- Achalasia is the best understood example of an esophageal motility disorder and characterized by esophageal aperistalsis and impaired relaxation of the lower esophageal sphincter.
- The histopathology of achalasia involves inflammation of the myenteric plexus of the esophagus with diminution of ganglion cells. Significant reduction in nitric oxide synthase containing neurons has been demonstrated using immunohistochemical staining.
- Autoimmune, neurodegenerative, and viral etiologies have been implicated in the pathogenesis of achalasia. However, the exact cause has yet to be elucidated.
- Pharmacologic studies in achalasia patients support the selective loss of inhibitory, nitrergic neurons with preservation of cholinergic innervation.
- Animal models that include genetically engineered mice with targeted disruption of the gene encoding for the neuronal form of nitric oxide synthase and pharmacologic administration of inhibitors of nitric oxide substantiate the role of nitrergic denervation in achalasia.
- Achalasia may be secondary to a wide range of secondary disorders including genetic syndromes, infectious diseases, neoplasm, and chronic inflammatory conditions.
Introduction
Sir Thomas Willis is credited with the first report of a patient with achalasia in 1674. Von Mikulicz in 1882 and Einhorn in 1888 hypothesized that the disease was due to the absence of opening of the cardia or "cardiospasm." Over the past three centuries, achalasia has emerged as an important model by which to understand the pathophysiology and therapy of motility disorders emanating from a defect in the enteric nervous system. It is the most extensively studied and readily treatable gastrointestinal motor disorder. This review discusses current concepts in achalasia with an emphasis on the pathophysiology and etiology of the disease. Specific secondary etiologies of achalasia are discussed that provide insight into mechanisms responsible for the neurodegeneration that characterizes the disorder. Diffuse esophageal spasm is also discussed, although there is a paucity of data regarding this condition.
Clinical Features of Achalasia
Achalasia occurs with an incidence of approximately 1:100,000 with an equal gender distribution.1 It occurs at all ages with an increase in incidence observed after the seventh decade. Dysphagia is the predominant symptom and it is typically accompanied by regurgitation. Upper endoscopy is often the first test used to evaluate patients with suspected achalasia and may detect esophageal dilatation with retained saliva or food. A barium esophagram can be highly suggestive of the diagnosis of achalasia, particularly when there is the combination of esophageal dilatation with retained food and barium and a smooth, tapered constriction of the gastroesophageal junction. Quantitative assessment of the degree of esophageal emptying of barium over time may increase the diagnostic sensitivity of the esophagram for achalasia and serves as a valuable means by which to follow patients response to therapy (Figure 1).2, 3 The test with the highest sensitivity in the diagnosis of achalasia is esophageal manometry. The defining manometric features of achalasia are aperistalsis of the distal esophagus and incomplete or absent lower esophageal sphincter (LES) relaxation (Figure 2). Additional supportive features include a hypertensive lower esophageal sphincter and low-amplitude esophageal body contractions. Preservation of proximal esophageal peristalsis can be seen in some cases without esophageal dilation (Figure 3a) but a pattern of complete esophageal aperistalsis is more common (Figure 3b).
Figure 1: Timed barium swallow.
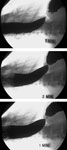
Following the ingestion of a fixed volume of barium, sequential radiographs are taken at 1, 2, and 5 minutes. The three panels demonstrate a lack of emptying with a fixed column of barium persisting at 5 minutes.
Figure 2: Esophageal manometric findings in achalasia.
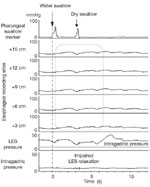
The tracing illustrates the findings in classic achalasia with esophageal body aperistalsis with low-amplitude simultaneous esophageal body contractions and failed relaxation of the lower esophageal sphincter.
Figure 3: Contour plot topographic analysis of esophageal motility in achalasia.

Topographic analysis is a method of axial data interpolation derived from computerized plotting of data from multiple, closely spaced, solid-state recording transducers. The interpolated pressure information is plotted as a two-dimensional contour plot in which pressure amplitude is coded by color. a: A normal esophageal study with propagation of the peristaltic wave and relaxation of the lower esophageal sphincter (LES). The upper esophageal sphincter is also depicted at the top of the panel, demonstrating a higher basal pressure and shorter relaxation phase. b: Study for a patient with achalasia demonstrating integrity of proximal esophageal peristalsis with mid and distal esophageal body aperistalsis (simultaneous contractions). Incomplete relaxation of the LES and elevated esophagastric pressure gradient are also demonstrated. c: A study from a patient with achalasia with complete esophageal aperistalsis and incomplete relaxation of the hypertensive LES. Although there is partial inhibition of the LES, the relaxation pressures exceed 40 mmHg. An esophagogastric pressure gradient is evident in the distal esophagus.
High-resolution manometry combined with topographic analysis is an emerging technique that offers potential advantages over conventional esophageal manometry.4 Using this technique, a threshold value of 8 to 10 mmHg for the mean residual pressure in a 3-second postdeglutitive interval distinguished achalasia patients from controls.5 Greater accuracy was achieved utilizing the transsphincteric pressure gradient during the 2- to 6-second post-swallow interval. Pressure gradients exceeding 5 mmHg had a sensitivity of 94% and specificity of 98% for detecting achalasia. Although an emerging methodology for investigative purposes, the advantages of high-resolution manometry with topography over conventional manometry for clinical practice are still being established. Figure 3 illustrates a contour plot topographic analysis of two patients with achalasia.
Although manometry is accepted as the "gold standard" for making the diagnosis of achalasia, heterogeneity does exist in the manometric presentation.6 The most commonly recognized variant of achalasia is known as "vigorous achalasia," variably defined by the presence of normal to high-amplitude simultaneous esophageal body contractions in the presence of a nonrelaxing LES (Figure 4). The amplitudes of preserved esophageal body contractions used to define vigorous achalasia have ranged from 37 mmHg to 60 mmHg. High-amplitude and long-duration esophageal body contractions have also been reported. These manometric features of vigorous achalasia overlap with diffuse esophageal spasm. Although vigorous achalasia may represent an early stage of achalasia, studies have failed to demonstrate differences in terms of clinical presentation in such patients including duration of disease or the occurrence of chest pain. Additional manometric variants of achalasia include rare patients with intact peristalsis through the more than 50% of the distal esophageal body and others with preservation of either deglutitive or transient LES relaxation (Figure 5).6, 7 In these reports, the diagnosis of achalasia was substantiated by demonstrating degeneration of myenteric neurons as well as the clinical response to disruption of the LES. The clinical significance of defining these variants of achalasia lies in the recognition that these sometimes confusing manometric findings are still consistent with achalasia when combined with additional data supportive of the diagnosis. The variants also provide clues to the pathophysiology of the disease.
Figure 4: Esophageal manometric findings in vigorous achalasia.

The recording is from a patient with vigorous achalasia demonstrating robust, simultaneous esophageal body contractions and failed relaxation of a hypertensive lower esophageal sphincter.
Figure 5: Esophageal manometric findings in achalasia variant with preserved LES relaxation.

The panel illustrates the findings in classic achalasia with esophageal body aperistalsis with low-amplitude simultaneous esophageal body contractions and apparent relaxation of the lower esophageal sphincter.
In addition to limitations in the sensitivity of the manometric features of achalasia, there also exist limitations in their specificity. Scleroderma shares many of the manometric features of achalasia with the exception of failed deglutitive relaxation of a hypertensive LES. This distinction becomes blurred in the setting of idiopathic achalasia with a low-normal LES basal pressure or treated achalasia. In addition, a prominent crural diaphragm contribution to LES basal pressure can be misinterpreted as the intrinsic basal LES pressure, and when combined with the presence of esophageal body aperistalsis of scleroderma esophagus, leads to a misdiagnosis of achalasia. Complicating matters, the esophageal manifestations of scleroderma may be the initial presentation in some patients with scleroderma. As mentioned, diffuse esophageal spasm has several manometric features that overlap with the vigorous form of achalasia. This overlap, combined with reports of progression of patients from esophageal spasm to achalasia, has led some authorities to question the existence of esophageal spasm as a distinct entity. Finally, a number of secondary causes of achalasia exist and are manometrically indistinguishable from idiopathic achalasia. The most clinically important secondary causes that need to be differentiated include pseudoachalasia as a consequence of neoplastic infiltration of the gastroesophageal junction and mechanical obstruction of the junction by a surgically created fundoplication. These and other secondary causes are discussed in a later section.
Pathophysiology of Achalasia
Esophageal Motility Abnormalities
Histopathology
Over the past 75 years, several pathologic studies have demonstrated the marked diminution of neurons from the myenteric plexus in achalasia.8, 9, 10, 11, 12 Illustrative of this is the large series by Goldblum's group13 in which complete absence of myenteric ganglion cells was demonstrated in 64% and marked reduction in 36% of the esophagi of 42 patients with achalasia who underwent esophagectomy. The same investigators also demonstrated a marked, T-cell predominant inflammatory infiltration of the myenteric plexus with fibrosis that was inversely correlated with the number of preserved ganglia.11 Recent studies have examined muscle biopsies from achalasia patients treated at an earlier stage of disease. These studies have detected intact, though a reduced number of, ganglion cells in achalasia patients having a shorter duration of symptoms and a nondilated esophagus,10, 14, 15 or preservation of esophageal contractile activity.11 The supposition is that myenteric inflammation occurs early in the natural history of achalasia and leads to aganglionosis and fibrosis. Figure 6 depicts a panel of varying degrees of myenteric inflammation and aganglionosis.
Figure 6: Histopathology of achalasia.

a: Normal myenteric plexus demonstrating multiple ganglion cells and minimal lymphocytic infiltration. b: Mild myenteric inflammation. There is mild lymphocytic inflammation, and ganglion cells can be identified. c: Moderate myenteric inflammation with lymphocytic infiltrate is present. Ganglion cells are absent. d: Severe myenteric inflammation with lymphocytes densely clustered within this myenteric plexus. Ganglion cells are absent. (Source: Hirano and Kahrilas112 with permission from Blackwell Publishing.)
Histologic examination of the smooth muscle of the esophagus of patients with achalasia have demonstrated a distinct abnormalities.10, 11, 16 Goldblum16 described hypertrophy as well as muscle degeneration of the muscularis propria and muscularis mucosae in the majority of 42 cases that underwent resection. Using high-frequency ultrasonography, Mittal et al.17 demonstrated a marked increase in both muscle wall thickness as well as cross-sectional area in achalasia patients. The increase in muscle mass was present in patients with and without esophageal dilation. The mechanism responsible for the muscle hypertrophy is unclear. Obstruction of esophageal outflow has been shown to result in secondary muscle hypertrophy in animal models.18, 19 In addition, the deficiency in nitric oxide that characterizes achalasia could also be responsible. Nitric oxide has an inhibitory effect on smooth muscle proliferation, and visceral smooth muscle hypertrophy has been described in neuronal nitric oxide synthase knockout mice.20
Interestingly, 52% of 42 specimens from esophageal resections for achalasia had eosinophilia of the muscularis propria. Tottrup et al.21 detected increased expression of eosinophilic cationic protein in achalasia, suggesting a possible pathogenic role for activated eosinophils in achalasia. Although eosinophils are not present in the esophagus of control specimens, no relationship between the increasingly recognized entity of eosinophilic esophagitis and achalasia has been established. Eosinophilic esophagitis is characterized by the finding of eosinophilic infiltration of the squamous mucosa, although cases of infiltration of the muscularis propria have been reported.22
Integrity of Cholinergic Innervation
A number of physiologic studies have uncovered an intact cholinergic innervation to the esophagus in achalasia. An in vitro study by Trounce et al.23 in 1957 demonstrated contractions of muscle strips from achalasia patients to the combination of the acetylcholinesterase inhibitor, eserine, and the ganglionic agonist, nicotine. Intact acetylcholinesterase activity of preserved ganglion cells in the lower segment of the esophagus of achalasia patients was described by Adams24 in 1961. The acetylcholinesterase inhibitor edrophonium chloride was later shown to significantly increase the LES pressures in patients with achalasia.25 These findings suggest that at least some postganglionic, cholinergic nerve endings remain intact. Further evidence in this regard came from a study looking at the effects of the anticholinergic agent atropine in patients with achalasia.26 This study demonstrated a 30% to 60% reduction in LES pressure with atropine in patients with achalasia. A similar reduction was found in a control group of healthy volunteers. Of note, however, is the fact that the residual pressure after atropine was significantly higher in the achalasia patients (17 mmHg) than in the normal subjects (5 mmHg).
Recently, botulinum toxin has been introduced as a novel treatment for achalasia. Botulinum toxin acts to inhibit the exocytosis of acetylcholine from cholinergic nerve endings. Most studies using botulinum toxin have found a significant symptomatic response rate. However, objective measures of response including LES pressure and esophageal emptying were modest and in some studies not significantly different from baseline values.27, 28 Similar to the studies using atropine, a significant residual LES pressure was observed following botulinum toxin, 25 mmHg in a study by Pasricha et al.28 and 20 mmHg in a study by Cuilliere et al.29 Therefore, the studies using atropine and botulinum toxin both support the concept of preservation of cholinergic nerves in patients with achalasia. Furthermore, they have provided evidence for a significant, noncholinergic component to LES basal pressure. It is likely that this residual pressure represents the myogenic contribution to LES tone. Heterogeneity in the response to botulinum toxin suggests a variable degree of cholinergic preservation of individual achalasia patients.
Loss of Inhibitory Innervation
Preservation of the excitatory, cholinergic innervation to the esophagus implies that the neuronal loss that characterizes achalasia may be selective for inhibitory neurons. Dodds et al.30 provided indirect evidence for this through the use of cholecystokinin, which has direct excitatory effects on smooth muscle as well as indirect inhibitory effects via postganglionic inhibitory neurons. In patients with achalasia, cholecystokinin induced LES contraction as opposed to the relaxation of the LES seen in control subjects, thereby providing evidence for impaired postganglionic inhibitory nerves. More recent evidence comes from in vitro studies looking at the responses of preparations of LES specimens from patients with achalasia. Circular muscles strips of the LES from normal subjects characteristically relax in response to electrical field stimulation through the activation of nitric oxide containing inhibitory neurons.31, 32 Paradoxically, LES strips from achalasia patients were found to contract in response to electrical field stimulation (Figure 7).31 Such findings can be readily explained by the absence of inhibitory neurons and presence of excitatory neurons.
Figure 7: In vitro study demonstrating the effects of electrical field stimulation (EFS) on circular muscle strips from the LES of control subjects (a) and patients with achalasia (b).

Electrical field stimulation activates enteric neurons, which results in relaxation of the LES that is unaffected by atropine (Atr) but abolished by tetrodotoxin (TTX). In the LES from an achalasia patient, ERS results in paradoxical contraction that is sensitive to both Atr and TTX. This experiment provides evidence for the loss of intrinsic inhibitory innervation with preservation of cholinergic excitatory innervation in achalasia. (Source: Tottrup et al.31 with permission from the BMJ Publishing Group.)
Evidence to support the concept of inhibitory neuronal loss came from both immunohistochemical and physiologic studies. Early studies postulated a defect in the adrenergic, inhibitory innervation of the esophagus.24 Vasoactive intestinal polypeptide (VIP) was also considered a candidate inhibitory neurotransmitter of the esophagus, and a reduction in VIP-containing neurons was demonstrated in achalasia patients.33, 34, 35 Following the discovery of nitric oxide, a number of studies have demonstrated the absence of nitric oxide synthase–containing neurons in LES specimens from patients with achalasia.6, 36, 37 Furthermore, recent experimental studies have shown that selective blockade of the inhibitory arm of the enteric innervation of the esophagus produces a manometric picture that closely mimics that of achalasia. Studies in both experimental animals and human subjects have shown that inhibition of nitric oxide synthase increases the resting tone of the LES and nearly abolishes LES relaxation (Figure 8a).32, 38, 39, 40 In the esophageal body, inhibition of nitric oxide synthase results in loss of the normal latency gradient manifest as simultaneous esophageal body contractions (Figure 8b). In healthy subjects, intravenously administered, recombinant hemoglobin, which inactivates nitric oxide, has been shown to produce simultaneous esophageal body contractions and failed LES relaxation (Figure 9).39 This motility pattern mimics the motility pattern of the vigorous form of achalasia (Figure 4). It lends credence to the model of achalasia that incorporates the selective loss of inhibitory and preservation of cholinergic enteric neural function (Figure 10b).
Figure 8: a: Effect of nitro-L-arginine methyl ester (L-NAME) on LES relaxation in the opossum in vivo.
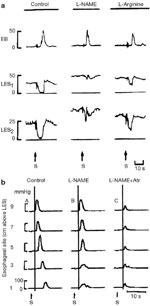
Control esophageal manometry illustrates an esophageal body peristaltic contraction and LES relaxation in two separate recording channels. Following the intravenous administration of L-NAME, a selective inhibitor of nitric oxide synthase, the latency prior to the esophageal contraction is shortened, basal LES pressure increased and LES relaxation is abolished. L-arginine, an antagonist of L-NAME, reverses the effects of L-NAME. (Source: Yamato et al.32 with permission from Elsevier.) b: Effect of L-NAME on esophageal peristalsis in the opossum in vivo. Control esophageal manometry (A) demonstrates an esophageal body peristaltic sequence with increasing latency period before each contraction that characterizes peristalsis. In the second sequence (B), the animal has been pretreated with L-NAME, a selective inhibitor of nitric oxide synthase, resulting in a loss of the latency gradient and hence simultaneous esophageal body contractions. Finally in the third panel (C), the animal was pretreated with both L-NAME and atropine (Atr), an antimuscarinic agent, resulting in simultaneous and low-amplitude esophageal contractions.
Figure 9: Effect of recombinant hemoglobin that inactivates nitric oxide on esophageal peristalsis in a human subject.
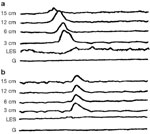
Control manometry (a) reveals a sequential peristaltic wave and normal LES relaxation in response to a swallow. Following an infusion of hemoglobin (b), deglutition produces simultaneous esophageal contraction and absent LES relaxation, mimicking the manometric profile of achalasia. (Source: Murray et al.39 with permission from American Gastroenterological Association.)
Figure 10: Pathophysiology of idiopathic achalasia.

Left: The normal condition where excitatory, cholinergic (Ach) motor neurons innervate the smooth muscle cells of the LES and contribute to the genesis of basal pressure of the LES (LESP). Inhibitory, nitric oxide (NO) motor neurons also act on the LES to produce the relaxation that accompanies a swallow. Middle: Achalasia resulting from the loss of inhibitory neurons. In this situation, the absence of NO motor neurons results in an elevation in the basal LESP and absence of swallow induced relaxation of the LES. Esophageal aperistalsis is defined by simultaneous esophageal body contractions. Right: Achalasia with complete loss of myenteric neurons. Here the basal LESP is below normal owing to the absent excitatory neurons, and swallow-induced relaxation is absent owing to the lack of inhibitory neurons. Esophageal aperistalsis is defined by the absence of esophageal body contractions. (Source: Hiranoand Kahrilas112 with permission from Blackwell Publishing.)
Pathologic studies that have examined esophageal resection specimens from achalasia patients undergoing esophagectomy likely select patients with long-standing or end-stage disease. The complete aganglionosis demonstrated in the majority of such patients may represent an end result of ongoing myenteric inflammation. Such descriptions support a second model of achalasia in which both the excitatory, cholinergic neurons and inhibitory, nitric oxide neurons are absent (Figure 10c). Under such circumstances, the functional obstruction of the gastroesophageal junction is caused by the residual myogenic tone of the LES. Complete absence of esophageal peristaltic activity is the result of the absent enteric neural innervation.
Although the nitric oxide–containing neurons may be lost in achalasia, the intracellular pathways by which nitric oxide acts remain intact in achalasia. This is evidenced by the efficacy of exogenously administered nitrates that act as nitric oxide donors in the treatment of achalasia. Furthermore, a study by Bortolotti et al.41 demonstrated a significant decrease in LES pressure following administration of sildenafil (Viagra). Sildenafil is an inhibitor of phosphodiesterase type 5 that breaks down cyclic guanosine monophosphate stimulated by nitric oxide. The efficacy of sildenafil in achalasia supports the integrity of the nitric oxide second messenger system in the smooth muscle of the LES in achalasia.
Extraesophageal Manifestations of Achalasia
Evidence supporting gastrointestinal functional abnormalities outside the esophagus is limited. Gastric functional abnormalities that include impaired proximal gastric accommodation, rapid emptying of liquids and decreased gastric acid secretion are consistent with an abnormality of either vagal or enteric innervation to the proximal stomach.15, 42, 43 Furthermore, a marked decrease in both ganglion cells and nitric oxide neurons has been demonstrated in the gastric body of achalasia patients, suggesting that the enteric neuropathy can extend beyond the esophagus.15, 44 However it should be noted that the abnormalities did not exist in all patients, and some studies have not detected impairment of gastric acid secretion or vagal function.42, 45 In the study by Csendes et al.,15 less than half of the achalasia patients had a decrement in ganglion cell concentration in the mid-stomach in contrast to esophageal aganglionosis that was present in over 90% of patients. In this same study of 34 achalasia patients, gastric emptying of solids was not significantly different from that in controls. Biopsies taken of the mid-jejunum and transverse colon showed a neuronal density that was similar to that in controls. Overall, pathologic and physiologic abnormalities in the stomach can be demonstrated in a subset of patients with achalasia but are rarely of clinical significance. Perturbations of gallbladder, sphincter of Oddi and small bowel motility have been reported but are uncommon and generally not of clinical significance.46, 47
Animal Models of Achalasia
Several animal models have been developed that have provided insight into the pathophysiology of achalasia. It should be emphasized that none of the animal models possesses all the pathophysiologic features of achalasia. Thus understanding the natural history and treatment of idiopathic achalasia using such models has been limited. The simplest model involves the surgical creation of a mechanical obstruction of the gastroesophageal junction. Little et al.48 created a feline model by fixing a 1-cm Gore-Tex band around the gastroesophageal (GE) junction. With this model, basal LES pressure increased slightly and a mild but significant impairment of deglutitive relaxation was achieved. Simultaneous esophageal body contractions were demonstrated in none of the animals preoperatively compared with 85% at 4 weeks. Although esophageal dilatation was demonstrated, no significant increase in muscularis thickness was demonstrated. The manometric changes were reversible following removal of the band.
Using a pressure cuff that applied variable degrees of obstruction of the GE junction in cats, Mittal et al.49 demonstrated that the esophageal body amplitudes and propagation were variably affected by both degree of distal obstruction as well as bolus volume. A similar model was created in the opossum by Tung's group.18, 19, 50 Both esophageal dilatation and an increase in thickness of the muscularis were demonstrated. Histologic study in these animals demonstrated hypertrophy of individual smooth muscle cells at both 4 and 8 weeks postoperative time points as well as some morphologic changes in the myenteric ganglia.50 In vitro muscle strip studies demonstrated reduced hyperpolarization in response to electrical stimulation in the hypertrophied muscle compared with controls suggesting a possible mechanism for the simultaneous contractions. A recent study examined the effects of more complete esophageal obstruction in the opossum that resulted in significant elevation in LES basal pressure and marked reduction of deglutitive relaxation from 99% to 28%. Degeneration of 5% to 20% of myenteric ganglion cells at 2 weeks following GEJ banding and 50% to 65% of myenteric ganglion cells at up to 6 weeks postoperatively were demonstrated.51 In the majority of animals, peristaltic function failed to recover after removal of the band. These experimental models lend credence to the possibility that esophageal aperistalsis can be secondary to distal esophageal obstruction as an alternative to a primary result of enteric neuropathy of the esophageal body.
Surgical vagotomy has also been examined as a model for achalasia. An achalasia-like syndrome was produced in dogs following electrolytic lesions of the medulla and bilateral vagotomy.52, 53 However the manometric and histologic findings varied from that observed in achalasia patients, leading to another investigation of the effects of cervical vagotomy in primates.54 In this study, only two of the seven monkeys developed a radiographic and manometric picture consistent with achalasia. Interestingly, the majority of animals demonstrated a significant reduction in the number of esophageal myenteric ganglion cells. Transection or cooling of the vagus nerve has also been shown to abolish primary peristalsis but leave secondary peristaltic function intact in the opossum.55, 56 These animal models demonstrate that vagotomy does not consistently produce an achalasia-like picture. Moreover, significant and lasting dysphagia is uncommonly observed following surgical vagotomy in humans. In a study of 96 patients who had undergone proximal gastric vagotomy for ulcer disease, only five patients developed transient albeit severe dysphagia with delayed transit of barium and incomplete LES relaxation.57
Gaumnitz et al.58 produced a model of achalasia in the opossum by injecting the gastroesophageal junction with a cationic surfactant that resulted in chemical denervation. Although these animals manometrically resembled achalasia with hypertension and failed relaxation of the LES, further study is needed to delineate whether the effects of the chemical injury are specific for enteric neurons rather than the result of a mechanical obstruction owing to injury to other structures of the gastroesophageal junction. If this model were substantiated, examination for secondary changes to the autonomic nervous system would be of great interest.
Most recently, genetically engineered mice with targeted disruption of the gene encoding the neuronal form of nitric oxide synthase have been studied.59 Hypertension of the LES was demonstrated as well as marked impairment of swallow-induced LES relaxation. The phenotype of this knockout mouse was dominated by marked gastric distention indicative of the importance of nitric oxide in gastric motility. The absence of significant esophageal dilatation may be explained by the predominance of striated muscle in the tubular esophagus of the mouse. Nevertheless, this model leads credence to the importance of inhibitory myenteric neural innervation in the pathogenesis of achalasia.
Sensory Function in Achalasia
Impaired Visceral Sensation
Although the pathophysiology of motor dysfunction has been extensively investigated, little is known about the integrity of esophageal sensory function in achalasia. Afferent innervation of the esophagus and conscious perception of sensation depends on vagal and spinal afferent fibers communicating with the central nervous system.60, 61, 62 Degeneration of the central, autonomic, or enteric nervous systems could lead to impaired visceral sensation in achalasia. Circumstantial evidence for such a functional impairment comes from the observation that patients with achalasia are poorly cognizant of retained food in the esophagus or presence of esophageal distention. Furthermore, uncontrolled studies have reported that achalasia patients have diminished perception of acid reflux events both before and after treatment of their achalasia.63
A limited number of investigations have examined esophageal sensation in achalasia. Two previous studies have evaluated visceral sensitivity using intraesophageal balloon distention.64, 65 Both studies found impairment of sensation in achalasia patients. However, these studies employed fixed-volume, latex balloons. As a result of the technique, the amount of pressure stimulus applied to the esophageal wall varied depending on the degree of dilatation present secondary to the underlying disease state. This method is of limited validity in achalasia patients, where esophageal dilatation is common. Rate et al.66 reported diminished esophageal sensory responses to electrical stimulation in a cohort of patients with varied esophageal motility disorders that included achalasia. Brackbill et al.67 recently investigated sensory function in achalasia utilizing a barostat device that maintains a constant pressure stimulus independent of luminal diameter. This study demonstrated significant differences in esophageal mechanosensitivity in achalasia patients who reported higher thresholds for painful, distention-induced sensation than in healthy controls. The same investigators also demonstrated decreased chemosensitivity in patients with achalasia using a modified Bernstein test. These studies support the concept of impaired esophageal sensitivity in achalasia but do not identify whether the defect is occurring at the level of the central, autonomic, or enteric nervous system. Central desensitization owing to the chronicity of the esophageal distention and chemical irritation by retained esophageal contents might also explain these observations.
Chest Pain
In contrast to the diminished esophageal visceral sensitivity, chest pain does occur in patients with achalasia and has been reported in 17% to 63% of patients. The mechanism for chest pain is unclear and it is likely that more than one mechanism is involved. Proposed etiologies include secondary or tertiary esophageal contractions, esophageal distention by retained food, and esophageal irritation by retained medications, food, and bacterial or fungal overgrowth. Inflammation within the esophageal wall could also be involved. A prospective study, however, found no association between the occurrence of chest pain and either manometric or radiographic abnormalities. In the same study, patients with chest pain were noted to be younger and had a shorter duration of symptoms compared to patients without pain, suggesting that esophageal visceral pain may be less common with increasing neurodegeneration.68 Many patients with achalasia report a history of episodes of intense substernal chest pain that improve or resolve over the course of their disease. Interestingly, treatment of achalasia had little impact on the reporting of chest pain, despite relief of dysphagia. A conflicting retrospective study reported chest pain in 44% of patients with achalasia undergoing Heller myotomy but no association between chest pain and either age or duration of symptoms.69 In addition, chest pain resolved in 84% of patients after Heller myotomy. Despite the retrospective nature of the study, this report does suggest that etiology of chest pain in achalasia is likely heterogeneous. It is likely that varied etiologies for chest pain account for the variations in both its prevalence and response to therapy.
Etiopathogenesis
Primary Achalasia
The etiology of primary achalasia remains unknown, although several hypotheses have been put forth including genetics, viral infection, autoimmunity, and neurodegeneration. Each hypothesis seeks to account for the loss of ganglia from the esophageal myenteric plexus, although it is likely that the various theories do not operate independently.
Genetics
Childhood and familial cases of achalasia are very uncommon and do not support an important genetic predisposition to primary achalasia.70, 71, 72 Several cases of siblings with achalasia have been reported, many of whom were born of consanguineous parents. Fewer than 10 cases of apparent vertical transmission of achalasia have been reported worldwide, and there is only one case report of monozygotic twins with achalasia.73 Allgrove's syndrome is an autosomal recessive disease that presents in children with associated features of alacrima, adrenal insufficiency, mental retardation, and autonomic and peripheral neuropathy. This disease is discussed in the section that follows on secondary forms of achalasia. An intriguing report noted coexistent Hirschsprung's disease and achalasia in two male siblings. Both siblings were diagnosed shortly after birth with achalasia requiring cardiomyotomy and both were subsequently diagnosed with Hirschsprung's disease with pathology of the rectum showing absent ganglion cells.74
Viral Hypothesis
A number of studies have implicated viral agents in the pathogenesis of achalasia. An infectious etiology seems plausible in light of the uniform age distribution of the incident cases of achalasia. Furthermore, Chagas' disease, discussed below, is an existing example of an infectious pathogen that can cause achalasia. A preliminary report noted a statistically significant increase in antibody titers against measles virus in patients with achalasia compared with controls.75 Although this study has not been substantiated, another study using DNA hybridization techniques found evidence of varicella-zoster virus in three of nine myotomy specimens from patients with achalasia but none of 20 control specimens.76 DNA probes for cytomegalovirus and herpes simplex type I were negative in both achalasics and controls. The herpes virus family was specifically targeted in this study given their neurotropic nature. Furthermore, the predilection of the herpes viruses for squamous epithelium as opposed to columnar epithelium makes this an attractive hypothesis. Such tissue selectivity could explain why achalasia involves only the esophagus while sparing the remainder of the gastrointestinal tract. More recent studies using more advanced methods including polymerase chain reaction techniques, however, failed to detect the presence of measles, herpes or human papilloma viruses in myotomy specimens of patients with achalasia.77, 78, 79 These negative studies do not exclude the possibility of either an alternate viral species or resolved viral infection with disappearance of the inciting viral pathogen from the host tissue as being the etiology of achalasia. Supporting this possibility is a recent study demonstrating immunoreactivity of inflammatory cells from patients with achalasia in response to viral antigens despite the inability of the investigators to detect the virus in tissue samples.80
Autoimmune Hypothesis
Early descriptions of inflammatory infiltration of the affected regions of the esophagus in achalasia led to speculation of an autoimmune pathogenesis. Inflammatory infiltration of the myenteric plexus was present in 100% of specimens from a histologic analysis of 42 achalasia esophagectomy specimens.11 Immunohistochemical staining characterized the infiltrative cells as T cells positive for CD3 and CD8.81 A significant eosinophilic infiltration has been demonstrated in some patients with achalasia.16, 21 An association between achalasia and class II histocompatability antigen has been described, specifically identifying a higher genotypic frequency of the human leukocyte antigen (HLA)-DQw1, DQA1*0101, DQA1*103, DQB1*0602, and DQB1*0603 alleles in achalasia patients compared with controls.82, 83, 84, 85 Class II antigen expression on myenteric neurons could be targeted as foreign antigens. Storch et al.86 demonstrated antibodies against myenteric plexus in serum of 37 of 58 patients with achalasia and in only four of 54 healthy controls. This study also failed to detect antibodies in the serum of patients with Hirschsprung's disease or esophageal cancer and in only one of 11 patients with peptic esophagitis. A second study detected serum antibodies against myenteric neurons in seven of 18 achalasia patients but not in healthy controls or reflux patients.87 The patients' antibodies bound to neurons in enteric plexuses from tissue sections of both the esophagus and intestine of rats. However, because the defect in primary achalasia is quite specific for the esophagus, the significance of a circulating antibody that targets not only esophageal but also intestinal neurons is unclear. In another recent study, positive immunostaining of the myenteric plexus of the esophagus and ileum of the guinea pig and mouse were detected in the serum samples of 23 out of 45 achalasia patients. However, a similar degree of immunostaining was demonstrated in the serum of eight of 16 patients with gastroesophageal reflux disease. This suggests that the antineuronal antibodies detected may represent nonspecific or secondary phenomena that do not play a causative role in the pathogenesis of achalasia.88
Neurodegenerative Hypothesis
Neurodegeneration is a third proposed etiology for primary achalasia. Loss of neurons within the dorsal vagal motor nucleus and degenerative changes of the vagal nerve fibers have been noted.10, 89 Experimental lesions of the brainstem and vagus nerves in animal models (see below) can produce esophageal motility abnormalities that resemble achalasia. Such findings led investigators to speculate that the site of primary involvement in achalasia was in the dorsal motor nucleus and vagus nerve and that the myenteric abnormalities were secondary. The majority of pathologic studies, however, have found that the predominant abnormalities exist within the myenteric plexus with marked diminution or complete absence of ganglion cells as well as intense inflammatory infiltration of the myenteric plexus.9, 11 Neural inflammation has not been described in other parts of the autonomic or central nervous system of achalasia patients arguing against these being the primary site of denervation. Furthermore, defects in vagal innervation would be expected to lead to prominent clinical abnormalities outside the esophagus including gastric emptying disorders, which are uncommonly seen in achalasia. A number of studies have looked for autonomic effects on gastric physiology and provided inconsistent results.15, 42, 45 Significant abnormalities in esophageal function are uncommon clinical manifestations in patients who have had vagal transections. Additional evidence to support the neurodegenerative hypothesis comes from the description of Lewy bodies, intracytoplasmic inclusions characteristically found in Parkinson's disease, in the myenteric plexus and dorsal motor nucleus of achalasia patients.90 It is likely that the neurodegenerative changes in achalasia are secondary to viral or autoimmune-mediated destruction of the enteric ganglia.
Secondary Achalasia
A number of disorders can result in a clinical presentation that manometrically and radiographically resembles primary achalasia (Table 1). Secondary achalasia can occur in a form that is isolated to the esophagus or part of a generalized motility disorder affecting other parts of the gastrointestinal tract.60 These secondary forms provide important insights into the pathophysiology of achalasia.

Allgrove's or Triple A Syndrome
Allgrove first reported the triple A or Allgrove's syndrome in 1978 when he described two pairs of siblings presenting with achalasia, alacrima, and adrenal insufficiency. Subsequent studies have characterized the disorder as an autosomal recessive disease with additional features that include peripheral neuropathy, autonomic neuropathy, and mental retardation. The majority of cases have presented in children less than 10 years old, with the morbidity owing to hypoglycemia and dysphagia. Pathologic studies have demonstrated absence of ganglion cells in the esophagus as well as atrophy of the adrenal cortical zona fasciculate and reticularis, and both axonal degeneration as well as nerve fiber loss of peripheral nerves. A study reported linkage of the gene for triple A syndrome to chromosome 12q13.91 Tullio-Pelet et al.92 reported mutations in a novel gene (AAAS) encoding a regulatory protein referred to as ALADIN (for alacrima-achalasia-adrenal insufficiency neurologic disorder). The ALADIN protein is believed to function in the normal development of both the peripheral and central nervous systems. It has been proposed that the ALADIN protein is involved in the regulation of nucleocytoplasmic transport that is essential to the maintenance and development of specific tissues that include the esophageal myenteric plexus. Thus Allgrove's syndrome, like Hirschsprung's disease, appears to be a genetic disorder leading to tissue-specific defects of the enteric nervous system.
Multiple Endocrine Neoplasia
Achalasia can also occur as part of motility disorders affecting multiple regions of the gastrointestinal tract. Esophageal dysmotility and achalasia have been described as part of multiple endocrine neoplasia (MEN) type 2B93 and von Recklinghausen's neurofibromatosis.94 Unlike the pathologic finding of aganglionosis that characterizes primary achalasia and most forms of secondary achalasia, these two disorders have hyperganglionosis or neuronal dysplasia of the myenteric plexus of the gastrointestinal tract. Recent molecular genetic studies have provided major advances in the understanding of MEN. Over 90% of patients with MEN-2 have been found to have mutations in the RET proto-oncogene.95 This gene is localized on chromosome 10q11.2 and encodes a receptor tyrosine kinase expressed on neural crest cells. The medullary thyroid carcinoma, pheochromocytoma, mucosal neuromas, and gastrointestinal neuronal dysplasia that develop as part of MEN-2 can be explained by the fact that these tissues are all derived from the neural crest during fetal development. Other hereditary forms of achalasia with associated generalized gastrointestinal motility disorders have been described and await molecular genetic characterization.60
Chagas' Disease
Chagas' disease is a parasitic infection caused by Trypanosoma cruzi that is endemic to regions of Central and South America and Mexico. T. cruzi is transmitted from person to person via the blood-sucking triatomine (reduviid) insects with 10% to 30% of infected individuals developing a chronic infection that presents years or even decades after initial infection. Although any portion of the gastrointestinal tract can be involved, the esophagus is most commonly affected, manifest as secondary achalasia in 7% to 10% of chronically infected individuals. Antibodies directed at targets within the myenteric plexus have been demonstrated in patients with Chagas' disease and achalasia.96 Circulating immunoglobulin G (IgG) antibodies recognizing the M2-muscarinic acetylcholine receptor were detected in a series of patients with achalasia secondary to Chagas' disease at much greater frequencies than found in Chagas' patients without achalasia, patients with idiopathic achalasia, and healthy controls. These investigators further demonstrated functional effects of the antibody in terms of an in vitro, muscarinic agonist-like activity on isolated rat esophageal muscle strips. The significance of this antibody in the clinical presentation of achalasia in Chagas' disease is unclear because the pathogenesis still involves destruction of the myenteric neurons. Moreover, Dantas et al.97 have reported greater impairment of the cholinergic pathway in patients with achalasia secondary to Chagas' disease compared with patients with idiopathic achalasia and healthy controls.
Paraneoplastic Syndrome
Cancer is an important cause of secondary achalasia. It can produce achalasia or an achalasia-like picture by one of three mechanisms. The first and most common is by direct mechanical obstruction of the distal esophagus. This is referred to as pseudoachalasia and has been described with a number of cancers (Table 1). Neoplastic cells can also invade the submucosa of the LES and thereby disrupt the myenteric neurons, resulting in an achalasia-like picture that can be missed on endoscopic examination. Finally, tumors remote from the distal esophagus can cause achalasia through a paraneoplastic syndrome.98 This is an autoimmune response in which the tumor expresses a neuronal antigen that the host recognizes as nonself. Activated T cells as well as plasma cell antibodies directed at the antigen act to retard the growth of the tumor but react with portions of the nervous system outside of the blood–brain barrier.99 Anti-Hu [also known as type 1 antineuronal nuclear autoantibody (ANNA-1)] recognizes proteins expressed in cancer tissue as well as neurons of the central, peripheral, autonomic, and enteric nervous systems.100 The paraneoplastic syndrome is most commonly seen with small cell lung cancer but has also been described in neuroblastoma and prostate cancer patients. The gastrointestinal manifestations associated with the anti-Hu paraneoplastic syndrome include achalasia, gastroparesis, and intestinal pseudo-obstruction. Encephalomyelitis, sensory neuropathy, and cerebellar degeneration have also been described. Importantly, the gastrointestinal manifestations can often precede the diagnosis of the cancer, and the presence of the paraneoplastic response may portend a better prognosis.101 Rare cases have been described with intestinal aganglionosis and anti-Hu antibodies in the absence of neoplasm.102
Diffuse Esophageal Spasm
Diffuse esophageal spasm (DES) is a rare condition first described by Osgood in 1889. The diagnosis is based on the finding of simultaneous esophageal body contractions (Figure 11). Radiologic barium studies demonstrate impaired esophageal emptying and the presence of simultaneous and tertiary contractions that result in the characteristic "corkscrew" or chain of beads configuration of the esophagus (Figure 12). How rapid esophageal contractions need to occur to be considered simultaneous was examined by Hewson et al.,103 who found that propagation velocities that exceed 6.25 cm/sec are associated with esophageal retention. Simultaneous esophageal contractions are not specific for diffuse esophageal spasm and have been reported in patients with diabetes, connective tissue disorders, alcoholism, and gastroesophageal reflux disease. Moreover, simultaneous contractions occur in 10% of swallows of healthy subjects,104 which has led to the requirement that at least 20% of swallows must be simultaneous for the diagnosis of DES. If every swallow sequence is simultaneous or if failed LES relaxation is noted, the diagnosis of achalasia needs to be strongly considered. Of note, motility recordings from a single patient may demonstrate manometric findings consistent with achalasia on one swallow and DES on another (Figure 13). High amplitude, repetitive, and long duration contractions may be present but are not considered necessary for the diagnosis.105 This observation has been challenged, and studies that have examined symptom correlation in patients with DES have reported a significant association between episodes of chest pain and prolonged duration, high-amplitude esophageal contractions.106, 107
Figure 11: Esophageal manometry in diffuse esophageal spasm.

Esophageal motility tracing from a patient with diffuse esophageal spasm demonstrating simultaneous contractions of the esophageal body with intact LES relaxation.
Figure 12: Radiographic examination of diffuse esophageal spasm.
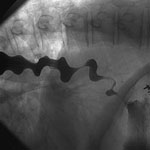
Barium swallow illustrating spiral or "corkscrew" deformity of the tubular esophagus caused by simultaneous, lumen-obliterated contractions of the circular muscle of the esophageal body.
Figure 13: Contour plot topographic analysis of esophageal motility and esophagram in diffuse esophageal spasm.

Plots show simultaneous standard manometry and contour plot topography from two water swallows from the same patient. a: High-amplitude, double-peaked and long duration propagated esophageal body contractions with partial LES relaxation. b: Simultaneous distal esophageal body contractions of normal amplitude and failed LES relaxation. c: Barium esophagram from the patient with a corkscrew-like morphology. This case illustrates the variable motility abnormalities that can be present at any one time with overlapping features of esophageal spasm and achalasia.
There are a number of similarities between DES and achalasia. Both conditions appear to result from a defect in the inhibitory neurotransmission of the esophagus, presumably at the level of the myenteric plexus. Failed deglutitive inhibition was demonstrated in patients with DES using paired swallows. The paired swallows at 5-second intervals generated a single peristaltic sequence in controls but two sets of esophageal contractions in patients with DES (Figure 14).108 In this same study, atropine was shown to reduce the frequency, amplitude, and duration of spontaneous contractions. Sifrim et al.109 created an artificial high-pressure zone in the esophageal body using a balloon and demonstrated absent deglutitive inhibition in patients with DES. Konturek et al.110 administered intravenous glyceryl trinitrate, a nitric oxide donor, to patients with DES and observed a dose-dependent elongation of the latency period as well as significant decrease in contraction duration. Administration of intravenous L-arginine, a nitric oxide precursor, did not have demonstrable effects. However, the study does support the hypothesized defect in nitric oxide innervation.
Figure 14: Failed deglutitive inhibition in diffuse esophageal spasm.
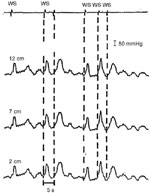
The tracing depicts simultaneous esophageal body contractions to repeated swallows at 5-second intervals. Normally, esophageal body contractile activity is inhibited with repetitive swallows with a peristaltic sequence following the final swallow. (Source: Behar and Biancani108 with permission from American Gastroenterological Association.)
Because of the rarity of cases of DES and the even less common need for surgical intervention, histopathologic data are few. Limited studies have demonstrated degeneration of vagal fibers, inflammatory infiltration of myenteric plexus, and hypertrophy of smooth muscle similar to findings described in achalasia. Furthermore, several cases have been reported in which transformation from diffuse esophageal spasm into achalasia has been observed.111
