

Key Points
- Esophageal mucosa consists of partially keratinized stratified squamous epithelium with three functional regions: stratum corneum, stratum spinosum, and stratum germinativum.
- Major esophageal defenses against injury by contact with an acidic refluxate are (1) luminal acid clearance, and (2) tissue resistance. Tissue resistance has three protective components: these designated as preepithelial, epithelial, and postepithelial defenses.
- The preepithelial defense consists of surface mucus and unstirred water layer within which bicarbonate ions are entrapped providing an alkaline microenvironment. The preepithelial defense in esophagus is weak relative to that of stomach and duodenum.
- The epithelial defense consists of the apical cell membranes and junctional complexes; these act to limit the diffusion of H+ from lumen to cell and intercellular space, respectively. In esophagitis, the junctional complexes are damaged, leading to increased H+ diffusion and dilation of the intercellular spaces.
- The postepithelial defense is provided by the acid buffering effects of HCO3- in cells and within the intracellular space. Blood supply is essential for the delivery of HCO3-. A Na+-dependent Cl-/HCO3- exchanger on the basolateral membrane of squamous cells provide a route for blood-derived HCO3- to enter the cell cytosol
- Cells in the stratum germinativum repair the damaged epithelium by two processes: restitution and replication. Epidermal growth factor (EGF) is a major promoter of cell replication and this can begin as early as 30 minutes after acid injury.
Introduction
Gastroesophageal (acid) reflux is in most subjects a benign physiologic process that occurs most commonly following meals. In these subjects, even repeated daily contact of the esophageal epithelium with acidic gastric contents does not produce damage to the tissue. This is because of the presence of two esophageal defenses: (1) the luminal acid clearance mechanisms, and (2) esophageal epithelial (tissue) resistance.1 The luminal clearance mechanisms come into play after reflux to assist with the rapid removal of both the bolus and the acidity of the refluxate. The bolus is removed by gravity and peristalsis, and acidity is negated by bicarbonate delivered to the lumen in swallowed saliva and secretions of the esophageal submucosal glands. Because luminal clearance is not immediate and may be delayed particularly during sleep, the esophagus must ultimately rely for protection on tissue resistance. This review defines tissue resistance, enumerates its components, and describes how they interact for protection of the epithelium against injury upon exposure to high levels of luminal acidity.
Tissue resistance is a working term that has been applied to all the structural and functional components of the esophageal mucosa that enable it to withstand contact with luminal content, especially acid, without damage.2 That the phenomenon exists is well documented in both animals and humans because it has been shown in vivo that even 3.5 hours of continuous perfusion of the rabbit esophagus with hydrochloric (HCl) acid, pH 2, or 30 minutes of continuous perfusion of the human esophagus with HCl, pH 1.1 (Bernstein test), does not produce damage to the epithelium.3, 4, 5 To understand how such protection is possible, it is helpful to view the components of tissue resistance as falling into three compartments—preepithelial, epithelial, and postepithelial—based on their anatomic relationship to the epithelium proper (Table 1). Despite this separation, it should be noted from the outset that these compartments do not stand alone but are dynamically intertwined, with each dependent on the others for its existence. Consequently, no one compartment is necessarily more valuable than the other for the defense of the tissue against injury upon contact with luminal content. This should become amply clear below in our discussion of how tissue resistance provides protection against injury from high levels of luminal acidity as encountered in vivo after a bout of gastroesophageal reflux.

Preepithelial Defense
The preepithelial factors comprising tissue resistance include a mucus layer, unstirred water layer, and bicarbonate ions entrapped within these layers. For the stomach and duodenum, the preepithelial defense is well developed, creating a substantial buffer zone between luminal content and the epithelial surface. This buffer zone for gastroduodenal epithelium is in part a function of the presence of a surface mucus layer and in part due to an aqueous bicarbonate-rich secretion from surface cells. Mucus protects by limiting access to the epithelium of large molecules, such as pepsin, though it is not effective in preventing luminal hydrogen ions (H+) access to the epithelium 6, 7 Nonetheless, bicarbonate secretion into the unstirred water layer creates a buffering zone that acts to neutralize back-diffusing H+, a phenomenon well documented by the fact that gastroduodenal epithelial surface pH remains at pH 5.0 to 7.0 under conditions where luminal acidity is as low as pH 2.0.8, 9, 10 The esophagus, unlike the stomach and duodenum, has a very limited surface buffer zone, with luminal pH of 2.0 yielding a surface pH of 2.0 to 3.010 (Figure 1). The reasons for this are that the esophagus lacks a mucus layer and its surface cells do not secrete bicarbonate ions.11, 12 The lack of a surface mucus layer is somewhat surprising given that the esophagus is bathed by swallowed saliva and secretions from its submucosal glands. This is because salivary and submucosal gland mucins are of the soluble type, for example, MUC5B, and are capable of lubrication but incapable of forming a fixed viscoelastic protective mucus layer.13, 14 Consequently, esophageal epithelium has greater exposure to luminal content, such as pepsin and bile salts, and more limited surface buffering of back-diffusing H+ than does the gastroduodenal epithelium. Indeed, this weakness of preepithelial defense in squamous-lined esophagus has been suggested as one reason why greater levels of acid suppression are needed for control of symptoms and for lesion healing in reflux esophagitis than in peptic ulcer disease—the former generally requiring the acid-suppressing potency of proton pump inhibitor PPI therapy and the latter requiring only the acid-suppressing potency of histamine-2 receptor antagonists.15 Despite the limited buffer capacity of the preepithelial defense in esophagus, it still has potential protective value. This is because the activity of the enzyme, pepsin, has been shown to lose its capacity to damage intact esophageal epithelium or isolated esophageal cells at a pH 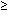 3.0.16 Consequently, even this modest level of surface buffering in esophagus can protect by negating the potential for more rapid and severe damage produced by the combination of acid-pepsin than by acid alone.
3.0.16 Consequently, even this modest level of surface buffering in esophagus can protect by negating the potential for more rapid and severe damage produced by the combination of acid-pepsin than by acid alone.
Figure 1: Preepithelial defense.
In gastric and duodenal epithelia, hydrogen ions (H+) must cross the mucus–unstirred water layer–bicarbonate barrier before contact can be made with the surface of the epithelium. Diffusion of pepsin, but not H+, is blocked by mucus; however, H+ can be neutralized by bicarbonate ions (HCO3-) residing in the unstirred water layer. In contrast to gastric and duodenal epithelia, the preepithelial defense in the esophagus is poorly developed, having a limited mucus–HCO3- barrier to buffer back diffusing H+. [Source: Orlando RC. Esophageal epithelial defense against acid injury. J Clin Gastroenterol 1991;13(suppl 2):S1, with permission.]
Epithelial Defense
When the preepithelial defense is bridged by luminal content, most typically back-diffusing H+, the esophagus relies for protection on the epithelium proper. Those factors within the epithelium that comprise this defense are listed in Table 1. In humans the esophagus is lined by a partially keratinized stratified squamous epithelium.17 This epithelium has the capacity to generate a transmural electrical potential difference in vivo, and this in large part is because of its capacity for active sodium ion (Na+) absorption across an epithelium of high electrical resistance (average 2000–3000 ohms.cm2).18, 19 Structurally, the esophageal epithelium has approximately 30 cell layers grouped into three functional regions known as the stratum corneum, stratum spinosum, and stratum germinativum. The stratum corneum comprises the uppermost seven to eight cell layers and these are primarily designed as a mechanical barrier for protection against luminal contents. Barrier function by these cell layers is provided by the apical cell membranes and adjacent intercellular junctional complexes, the latter consisting of tight junctions and zonula adherens (Figure 2). The tight junctions and zonula adherens have proteins such as occludin and claudins for the former and e-cadherin for the latter that bridge the space between neighboring cells, and below which is a glycoprotein-rich matrix within the intercellular space.20, 21, 22, 23 One component of the barrier function of the junctional complex in esophageal epithelium is calcium-dependent. This was established experimentally by showing that removal of calcium from the bathing solution results in marked reduction in transepithelial resistance and coincident increase in paracellular permeability. Moreover, this investigation indicated that the calcium-dependent component of junctional resistance was due to homophilic binding between molecules of e-cadherin from adjacent cells.24 Together the apical cell membranes and junctional complexes contribute to protection by limiting both the types and quantities of ions and molecules that can directly diffuse from the lumen into the cell or from the lumen into the intercellular space, respectively.
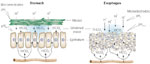
Figure 2: Epithelial defense.
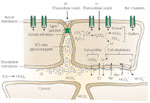
Some of the recognized epithelial defenses against acid injury are illustrated. Structural barriers to hydrogen ion (H+) diffusion include the apical cell membrane and intercellular junctional complex. Functional components include intracellular buffering by negatively charged proteins and bicarbonate ion (HCO3-) and H+ extrusion processes (Na+/H+ exchange and Na+-dependent Cl-/HCO3- exchange) for regulation of intracellular pH. Na+ = sodium ions. (Source: Modified from Orlando RC, Dobrucali AM. In: Feldman M, Orlando RC, eds. Atlas of Esophageal Diseases, 2nd ed. Philadelphia: Current Medicine, 2002: 99, with permission.)
For luminal acid to destroy esophageal epithelial cells, H+ must first cross the cell membrane in sufficient quantities to lower cytosolic pH, and second must sustain an acidic cytosolic pH long enough for it to destroy such vital cell processes as cell respiration. To do this H+ must find a route to the cell cytosol. One such route is directly across the apical cell membrane (Figure 2), a route that is very limited in opportunities based on its composition as a highly hydrophobic lipid bilayer. Yet the apical cell membrane has one pathway that seems ideally suited for H+ entry from lumen to cytosol and that is via the presence of apical membrane cation channels. This is made even more appealing given that these cation channels are "nonselective" in that they permit almost equal movement of Na+, K+, and Li+ ions through them.25 However, when surface cells in esophageal epithelium were impaled with pH-sensitive microelectrodes, lowering luminal acidity to as low as pH 2.0 resulted in little or no change in cytosolic pH.26 One reason for this lack of response is that the esophageal cation channels are regulated by acidic pH, with high levels of luminal acidity inhibiting channel activity so that neither Na+ nor H+ can traverse it.27 Consequently, acid regulation of the cation channel represents another means of intrinsic defense by which the epithelium protects itself from injury at high levels of luminal acidity.
An alternative route for H+ entry into the cell cytosol is across the junctional complex into the intercellular space and from the intercellular space across the basolateral membrane (Figure 2). This pathway has appeal in that the first change observed in the epithelium when luminal pH is lowered to noxious levels, that is, pH <2.0, is a drop in transepithelial electrical resistance. Because electrical resistance in esophageal epithelium is a parameter that primarily reflects changes to the junctions, this decline reflects an increase in paracellular permeability.28, 29 Moreover, the morphologic counterpart to this increase in junctional permeability is the development of dilated intercellular spaces within squamous epithelium.20, 30 Dilated intercellular spaces is an early change observed in acid exposed esophageal epithelium and is now recognized as an early hallmark of both erosive and nonerosive reflux disease31, 32, 33 (Figure 3). Further, because the sensory neurons in esophageal epithelium can extend within the intercellular spaces to within three cell layers of the lumen, the increase in paracellular permeability would increase H+ access to afferent neurons (nociceptors). Consequently, these events provide a plausible explanation for the development of heartburn in nonerosive reflux disease34, 35 (Figure 4). Despite the development of heartburn due to acid leak into the intercellular space, most subjects with heartburn do not develop erosive esophagitis. This indicates the presence of additional mechanisms for esophageal protection beyond the apical membrane-junctional complex that serve as physical barriers. Within the epithelium are in fact a number of functional defenses, and these include intracellular and extracellular buffers for H+, intracellular pH (pHi) regulation, and mechanisms for tissue repair (Table 1). Buffers for back-diffusing H+ include phosphates, proteins, and HCO3-, the latter generated intracellularly by the action of carbonic anhydrase and extracellularly by diffusion from the blood supply (Figure 2). The importance of buffers, particularly HCO3- is evident in that depletion of HCO3- experimentally results in cell necrosis at levels of luminal acidity that would otherwise not produce this outcome.36 Buffers initially work to prevent cell necrosis by preventing intercellular acidification from H+ diffusing across the junctions. This is the case because intercellular acidification is a prerequisite for cell acidification. Cell acidification occurs with intercellular acidification because the now acid-exposed basolateral cell membrane is more acid permeable than the apical cell membrane of esophageal cells. This is due to the presence within the basolateral membrane of a disulfonic stilbene-sensitive, (Na-independent) Cl-/HCO3- exchanger.36, 37 Therefore, when the intercellular space is acidic, the Na-independent, Cl-/HCO3- exchanger operates to acidify the cell cytosol by exchanging intracellular HCO3- for extracellular Cl-, an action equivalent to the absorption of HCl. This is supported experimentally by the ability of pharmacologic blockade of the Na-independent, Cl-/HCO3- exchanger to prevent in vitro acid-induced cell necrosis in esophageal epithelium.36, 37
Figure 3: Transmission electron micrographs of intercellular spaces.
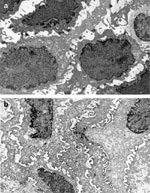
The electron micrographs show the differences in size of the intercellular spaces for normal (a) and nonerosive acid-damaged (b) esophageal epithelia.  9000. (Source: Barlow and Orlando,35 with permission from American Gastroenterological Association.)
9000. (Source: Barlow and Orlando,35 with permission from American Gastroenterological Association.)
Figure 4: Diffusion of refluxed gastric acid (H+) into the intercellular space.
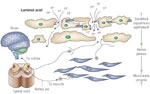
The presence of abnormal tissue resistance, demonstrated by defects within the intercellular junctional complex between cells of the surface layers of esophageal (stratified squamous) epithelium, is shown to enable the ready diffusion of refluxed gastric acid (H+) into the intercellular space. Within this space, it encounters and activates chemosensitive nociceptors whose signals are transmitted via the spinal cord to the brain for symptom (heartburn) perception. Activation of the same nociceptors is also capable of initiating a short reflex arc to esophageal (longitudinal) smooth muscle as means of precipitating a sustained esophageal contraction. (Source: Barlow and Orlando,35 with permission from American Gastroenterological Association.)
In addition to protection against intercellular acidification, intracellular buffers, especially HCO3- generated by the action of carbonic anhydrase, also protect against acidification of the cell cytosol.38 In primary cultured esophageal epithelial cells, it has been shown in vitro that they have the capacity to buffer the equivalent of  39 mmol of H+ at a pHi of 7.1.39, 40 Under conditions where acid entry exceeds the buffer capacity, pHi falls to acidic levels. At acidic pHi, the esophageal epithelium has available two acid-extruding mechanisms to prevent cell injury and death: an amiloride-sensitive Na+/H+ exchanger (NHE-1 isotype), and a disulfonic stilbene-sensitive, Na+-dependent Cl-/HCO3- exchanger.39, 40, 41, 42 The latter mechanism, however, is only operative when there is sufficient extracellular HCO3- for exchange for intracellular Cl- as occurs when pHi declines due to production of excess intracellular acidic metabolites. In this latter case, there is sufficient extracellular HCO3- to exchange for Cl- to raise pHi. However, when pHi declines because of acidification of the intercellular space, the Na+-dependent Cl-/HCO3- exchanger is inhibited, resulting in greater dependence on the action of the Na+/H+ exchanger for restoration of pHi to neutrality, by the exchange of intracellular H+ for extracellular Na+.
39 mmol of H+ at a pHi of 7.1.39, 40 Under conditions where acid entry exceeds the buffer capacity, pHi falls to acidic levels. At acidic pHi, the esophageal epithelium has available two acid-extruding mechanisms to prevent cell injury and death: an amiloride-sensitive Na+/H+ exchanger (NHE-1 isotype), and a disulfonic stilbene-sensitive, Na+-dependent Cl-/HCO3- exchanger.39, 40, 41, 42 The latter mechanism, however, is only operative when there is sufficient extracellular HCO3- for exchange for intracellular Cl- as occurs when pHi declines due to production of excess intracellular acidic metabolites. In this latter case, there is sufficient extracellular HCO3- to exchange for Cl- to raise pHi. However, when pHi declines because of acidification of the intercellular space, the Na+-dependent Cl-/HCO3- exchanger is inhibited, resulting in greater dependence on the action of the Na+/H+ exchanger for restoration of pHi to neutrality, by the exchange of intracellular H+ for extracellular Na+.
Postepithelial Defense
The blood supply is the postepithelial defense for the esophageal epithelium. It provides via its capillaries a steady supply of nutrients and oxygen for cell functions and repair, and removes metabolic wastes such as CO2 and acid. A key component of the blood supply in protection against acid injury is as a supply route for HCO3- delivery to the intercellular space. The HCO3- within the intercellular space serves both a buffering action there and within the cell cytosol—the latter enriched in HCO3- by the action of the Na+-dependent Cl-/HCO3- exchanger. Notably, the blood supply and its capacity to deliver HCO3- is a dynamic and not a static process so that under conditions of increasing luminal acidity, esophageal blood flow increases, and some of the known mediators of this increase are through the release within the tissue of histamine, nitric oxide, and calcitonin gene-related peptide.43, 44, 45, 46
Under conditions where acidification of the cytosol is prolonged, cell injury occurs, and this is reflected initially by cell swelling. Cell swelling occurs because of an acid-induced increase in intracellular Ca2+, which in turn activates a membrane NaK2Cl cotransporter. The NaK2Cl cotransporter enables high concentrations of Na+, K+, and Cl- to enter the cell at a time when such transporting mechanisms as the Na+-extruding, NaK–adenosine triphosphatase (ATPase), and K+-extruding K+ channels are inhibited by the low pH of the environment. Consequently, there is an increase in ion uptake and decrease in ion extrusion from the cell that results in an osmotic gradient that favors water uptake into the cell and thus cell edema.47, 48 Because cell edema effectively reduces the concentration of H+ within the cell, cell edema in a sense is a form of epithelial defense against excess acidity; that is, the cell sacrifices volume regulation for improved control (less acidic) of pHi.
Epithelial Repair
When low pHi is sufficiently prolonged to produce cell necrosis, defense of the epithelium as a whole becomes paramount, and this entails calling on strategies for repair, namely epithelial restitution and epithelial replication. For either of these processes to be effective, the cells of the lower layers, that is, stratum germinativum, must be preserved because they are the only ones that appear capable of restitution or replication.49, 50, 51, 52, 53 Restitution restores epithelial integrity by migration of healthy cells adjacent to an injury over the denuded basement membrane. Because this occurs without the need for protein or DNA synthesis, the process is rapid, and restitution has been shown to be stimulated by a variety of growth factors including hepatocyte growth factor, insulin-like growth factor, and epidermal growth factor (EGF).54, 55 Restitution, however, appears limited to the stratum germinativum because rapid recovery of moderate acid injury to the surface layers (evident by decline in electrical resistance in using chambered epithelium) has not been observed to occur despite monitoring for several hours.36 This may in part be due to the fact that restitution is readily inhibited at an acidic pHs of 3.0 to 6.5.54 Consequently, replication, not restitution, appears to be the major reparative defense for acid-injured esophageal epithelium. Replication in fact has been shown to begin within 30 minutes after esophageal acid exposure and is in evidence histologically by the presence of basal cell hyperplasia in esophageal biopsies of subjects with nonerosive reflux disease56, 57, 58, 59 (Figure 5). It is noteworthy that one stimulus for replication is EGF, a protein delivered to the esophageal lumen in swallowed saliva and secretions from the esophageal submucosal glands. For replication, luminal EGF appears to gain access to the EGF receptors on the basal cells in the stratum germinativum by diffusion across the acid-damaged junctional complex and through the intercellular space.30 Because the cell turnover rate in esophageal epithelium is 5 to 8 days,51, 52 repair in the absence of ongoing injury is theoretically possible in 2 to 4 days. Alternatively, when the rate of injury outpaces the rate of repair, nonerosive acid damage progresses to erosive esophagitis. Such a scenario is evident experimentally, though it appears relatively infrequent in patients with gastroesophageal reflux disease (GERD). The reason for this remains unknown but may lie in the type and extent of the injury, and the rate and capacity for repair or the type of inflammatory reaction.
Figure 5: a: Normal esophageal suction biopsy from a healthy subject without esophagitis.
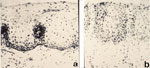
Basal zone thickness is approximately 10% of total epithelial thickness; papillae extend approximately one-half the distance to the epithelial surface. b: Abnormal suction biopsy from a subject with symptomatic reflux. Basal zone thickness is approximately 35% of total epithelial thickness; papillae extend over two thirds of the distance to the epithelial surface. BZ, basal zone, SZ, stratified zone; P, papillae; LP, lamina propria. Hematoxylin and eosin 170. (Source: Ismail-Beigi et al.,59 with permission from American Gastroenterological Association.)
Conclusion
The esophageal defenses described herein are an interdependent complex of structural and functional elements designed to provide the epithelium proper with protection during contact with noxious luminal substances, most notably refluxed acid and pepsin. These defenses are the final pathway and critical determinant of whether or not repeated episodes of reflux result in disease. Successful protection against acid-peptic exposure means that the subject remains asymptomatic and thus unaware of the events. Alternatively, failure of protection leads to epithelial damage—damage that when progressive translates into symptoms (heartburn) and signs of GERD. The damage that occurs, however, is not irreversible because the mucosal defenses include within their repertoire reparative elements capable of restoring health and integrity to the epithelium. The factors that ultimately govern whether health or disease prevail in the battle between the mucosal defenses and the refluxate remain poorly defined and an area of intense investigation.
