
Abstract
Purpose:
Diagnostic exome sequencing (DES) is now a commonly ordered test for individuals with undiagnosed genetic disorders. In addition to providing a diagnosis for characterized diseases, exome sequencing has the capacity to uncover novel candidate genes for disease.
Methods:
Family-based DES included analysis of both characterized and novel genetic etiologies. To evaluate candidate genes for disease in the clinical setting, we developed a systematic, rule-based classification schema.
Results:
Testing identified a candidate gene among 7.7% (72/934) of patients referred for DES; 37 (4.0%) and 35 (3.7%) of the genes received evidence scores of “candidate” and “suspected candidate,” respectively. A total of 71 independent candidate genes were reported among the 72 patients, and 38% (27/71) were subsequently corroborated in the peer-reviewed literature. This rate of corroboration increased to 51.9% (27/52) among patients whose gene was reported at least 12 months previously.
Conclusions:
Herein, we provide transparent, comprehensive, and standardized scoring criteria for the clinical reporting of candidate genes. These results demonstrate that DES is an integral tool for genetic diagnosis, especially for elucidating the molecular basis for both characterized and novel candidate genetic etiologies. Gene discoveries also advance the understanding of normal human biology and more common diseases.
Genet Med 19 2, 224–235.
Similar content being viewed by others
Introduction
Rare diseases affect 25–30 million people in the United States, 75% of whom are children, and 25% of pediatric inpatient admissions are due to these diseases.1,2 Although between 6,000 and 7,000 rare diseases with suspected genetic etiologies have been described (Orphanet), the responsible gene has been discovered for only half (3,500).3 It is estimated that mutations in up to 15,000 human genes are likely to lead to rare diseases;4 however, only between ~3,500 and ~7,000 genes have been implicated in some way with disease (Human Gene Mutation Database, OMIM).4,5
Dramatic advances in DNA sequencing technology over the past decade rapidly translated to fundamental changes in clinical care in at least two ways: by making large-scale genetic testing more affordable, and by accelerating the rate of scientific discovery of gene–disease associations, thereby increasing our ability to diagnose patients. Exome sequencing serves a dual role as a diagnostic and a discovery tool,6,7,8 and single case reports of patients in whom a candidate gene is identified on a clinical basis are becoming abundant.7,9,10,11,12 The first report of exome sequencing successfully identifying a disease gene was in 2010 (ref. 13), and within just 2 years of this achievement, at least 100 additional genes were characterized as causing disease.14 The discovery of new disease-causing genes is projected to be the next great revolution in molecular genetics.14 Since 2010 there has been a marked acceleration in the number of diseases for which the molecular basis is known, and it is estimated that all Mendelian disease genes will be uncovered by the year 2020 (ref. 3). As an illustration of the fast pace of disease-gene discoveries, we recently reported that among patients who underwent clinical diagnostic exome sequencing (DES), 23% of positive findings were within genes that were clinically characterized within the past 2 years.15
In a recent publication, clinicians from one clinic felt that candidate genes from DES were reported without stringent criteria and ultimately disagreed with the genes’ clinical relevance based on subsequent review of the literature, model systems, and patients’ clinical features.7 Therefore, it is critical that clinical laboratories develop a standardized, in-depth, and transparent system for scoring potential candidate genes for clinical reporting.
In this report, we describe the process for evaluating candidate genes for clinical reporting and provide the rate of candidate-gene reports among the 1,500 unselected consecutive cases that were referred to our laboratory for DES. Our results demonstrate that DES is not only an integral tool for genetic diagnosis but is particularly useful for elucidating the molecular etiology of genetic diseases.
Materials and Methods
Terminology
-
Uncharacterized gene–disease relationship: A gene–disease relationship and/or mechanism not previously proposed or with limited evidence based on Ambry’s clinical validity assessment criteria, derived from the ClinGen criteria (http://www.clinicalgenome.org/knowledge-curation/gene-curation/clinical-validity-classifications).
-
Characterized gene–disease relationship: A disease or phenotype whose underlying molecular etiology is established with at least a moderate level of evidence based on clinical validity assessment.
-
Characterized Mendelian disease gene: A gene known to underlie at least one Mendelian genetic condition.
-
Uncharacterized Mendelian disease gene: A gene that is not currently known to underlie a Mendelian genetic condition.
-
Candidate-gene criteria for clinical reporting: Criteria scheme to evaluate the level of available evidence to propose candidate gene–disease relationships ( Figure 1 ).
Figure 1 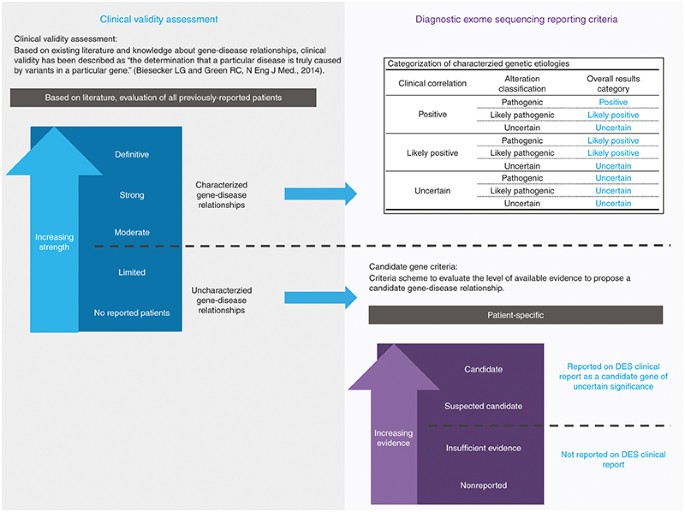
Clinical validity assessment versus candidate-gene criteria. Clinical validity assessment uses the peer-reviewed literature about gene–disease relationships along with previously reported patients to determine the level of existing evidence that a particular disease is truly caused by alterations in a particular gene. Candidate-gene criteria, which are patient specific, are used to determine the level of available experimental evidence available to propose a candidate gene–disease relationship.
-
Clinical validity: Based on the existing literature and knowledge about gene–disease relationships, clinical validity is the determination that a particular disease is truly caused by pathogenic variants in a particular gene ( Figure 1 ).
-
Whole-exome sequencing (WES): Library capture and sequencing of virtually all the coding exons within the genome. WES is a generic term that includes the collective of research, diagnostic, and clinical exome sequencing.
-
Diagnostic exome sequencing (DES): WES and computational analysis performed for diagnostic purposes for a single patient.

Patients/study population
Patients were ascertained sequentially through clinical samples sent to Ambry Genetics Laboratory for DES beginning in September 2011. Results summarized herein include those reported through November 2015. Clinicians were encouraged to refer multiple family members along with the proband for testing for family-centered exome sequencing and analysis, which included trio WES (generally the parents and proband), cosegregation analysis of candidate alterations using all informative family members, and computational analysis using family history inheritance-based filtering.
All patient identifiers covered by the Health Insurance Portability and Accountability Act were removed. Solutions Institutional Review Board determined the study to be exempt from the Office for Human Research Protections Regulations for the Protection of Human Subjects (45 CFR 46) under category 4. Retrospective analysis of anonymized data exempted the study from the requirement to obtain consent from patients.
Test options
For certain cases from September 2011 to April 2014, analysis of uncharacterized genes was not completed based on the test selected by the ordering clinician. Since April 2014, analysis of uncharacterized genes was performed on all informative trios that were negative for a reportable finding in a characterized gene. In all, analysis of uncharacterized genes was completed for 934 cases among a cohort of 1,500 probands.
Whole-exome sequencing
Exome library preparation, sequencing, and bioinformatics were performed as previously described.15 Briefly, samples were prepared using either the SureSelect Target Enrichment System (Agilent Technologies, Santa Clara, CA), SeqCap EZ VCRome 2.0 (Roche NimbleGen, Massion, WI),16 or the IDT xGen Exome Research Panel V1.0 (Integrated DNA Technologies, Coralville, IA) and sequenced using paired-end, 100- or 150-cycle chemistry on the Illumina HiSeq or NextSeq (Illumina, San Diego, CA). Stepwise filtering included the removal of common single-nucleotide polymorphisms, intergenic and 3′/5′ untranslated region variants, intronic variants outside ±2, and synonymous variants (other than potential splice-related synonymous changes at the first and last positions of exons). However, alterations classified as pathogenic or likely pathogenic based on Ambry’s variant classification schema as well as alterations with a Human Gene Mutation Database identifier were protected from the aforementioned filtering. Identified candidate alterations were confirmed using automated fluorescence dideoxy sequencing.
Clinical validity assessment and gene classification
Genes were classified as either uncharacterized or characterized Mendelian disease-causing genes based on Ambry’s clinical validity assessment criteria (data in preparation) ( Figure 1 ). Briefly, the assessment is inspired by the ClinGen clinical validity assessment criteria (http://www.clinicalgenome.org/knowledge-curation/gene-curation/clinical-validity-classifications), which scores evidence of gene–disease relationships using a tiered system: definitive, strong, moderate, limited, no reported evidence, and conflicting evidence reported. A team of scientists reviews the recently published peer-reviewed literature and assesses clinical validity daily. A gene is classified as characterized if there exists at least one gene–disease relationship with evidence that is moderate or stronger. Genes that have gene–disease relationships that are at most of limited evidence are classified as uncharacterized Mendelian disease genes. Gene–disease relationships classified as “limited” by clinical validity assessment have been reported in at least one patient; however, for clinical reporting purposes, the level of evidence is not sufficient to be considered “characterized”.
Reporting of primary results
Results were analyzed and reported as previously described.15 A candidate gene was reported if significant supporting evidence was found. Reports with candidate-gene findings, which were always interpreted as uncertain findings, fell under one of two categories: (i) “candidate” or (ii) “suspected candidate.” Alterations in genes with insufficient evidence to support likely clinical relevance were provided as a supplement to the report as “uncharacterized genes with an insufficient level of evidence.” All exome reportable findings are submitted to ClinVar, and alterations classified as “candidates,” “suspected candidates,” and alterations in uncharacterized genes classified as “insufficient evidence” are submitted to GeneMatcher.
Candidate-gene criteria
Classification of candidate genes was based on the criteria listed in Table 1 . All alterations were classified according to Ambry’s clinical variant classification scheme, which includes special classification considerations for alterations involved in candidate genes (http://www.ambrygen.com/sites/default/files/Reclassification%20Chart.pdf).17 The variant classification scheme for candidate genes focuses on evidence that the alteration is damaging to the protein function rather than pathogenic for disease. Per the criteria, alterations in candidate genes cannot meet the criteria to be classified as a “pathogenic mutation.” Alterations in uncharacterized genes with potentially reduced penetrance, variable expressivity, or potential mosaicism are not detected and/or evaluated. The candidate-gene criteria use a combination of the alteration classification along with supporting experimental data. To report a gene as a “candidate” or “suspected candidate,” a significant amount of experimental data that support the phenotype in question needs to be available. A candidate or suspected candidate cannot be proposed based on the potential deleterious nature of an alteration alone, given that healthy individuals are expected to carry ~300 rare,18 31 deleterious,4 and an average of 1 de novo19 protein-disrupting variant per exome. The candidate-gene criteria scoring system uses a combination of weighted evidence that includes previously reported patients, animal models, human microdeletion/duplication syndromes that include the gene of interest, gene function and expression profiles, and colocalization or interaction with the products of genes known to cause similar phenotypes. The evidence criteria are categorized into different weight levels (“A,” “B,” and “C”), in order of decreasing strength, determined by a team of molecular geneticists and inspired by previously published recommendations.20,21 No single piece of evidence can be used to collect points in more than one category or criterion. The criteria are then combined and assessed using the scoring rules defined in Table 1 to reach the ultimate category of “candidate,” “suspected candidate,” “insufficient evidence,” or “nonreported.”
Evidence components of the candidate-gene criteria
-
Previously reported patients: As described above, our clinical validity assessment criteria classify genes as uncharacterized Mendelian disease genes when gene–disease relationships are at most limited. A gene–disease relationship categorized as limited has fewer than three reported pathogenic alterations in unrelated individuals, and, in general, the gene–disease relationship has already been proposed in the literature, but insufficient supportive evidence has emerged to provide a stronger clinical validity assessment. Based on the candidate-gene criteria, if a gene–disease relationship previously received a clinical validity assessment score of limited and if the proposed disease phenotype was highly consistent with the patient’s phenotype, an “A” point is achieved and is therefore reported as a candidate.
-
Microdeletions/copy-number-variation syndromes that include the gene of interest: If the gene of interest is located within a well-defined critical gene region of an established microdeletion/copy-number-variation syndrome that has phenotypic features that are highly consistent with the patient’s reported phenotype, A-level evidence is achieved and the gene is reported as a candidate. However, if the gene of interest is simply located within a microdeletion/duplication syndrome that has been described in multiple patients with features consistent with the phenotype, a point within C-level evidence is achieved.
-
In vivo animal models: Observation of an alteration in the gene or gene ortholog of interest within a model organism with a highly specific phenotype is among the strongest nonhuman evidence available.20,21 B-level evidence is achieved when an in vivo model organism and the evaluated patient both have specific overlapping and uncommon phenotypes in the presence of variants that were expected to exert their effects via the same genetic mechanism (e.g., gain of function or loss of function). For instance, a patient with a heterozygous loss of function variant (nonsense, frameshift, splice) is compared only with a heterozygous knockout mouse model, not with a homozygous knockout, or with any mouse model carrying a missense variant unless it was experimentally demonstrated that the missense variant was truly loss of function. If the model organism either has a mutation with an inconsistent genetic mechanism compared with the patient or only a moderately consistent phenotype, C-level evidence is achieved.
-
In vitro studies: For loss of function alterations, when a gene disruption experiment produces a phenotype supportive of the proposed gene–disease relationship, C-level evidence is achieved. Demonstration that the phenotype can be rescued by complementation in a cellular assay can often be considered even stronger evidence than model systems;21 in such cases B-level evidence is achieved.
-
Protein family, colocalization, or interaction with genes known to cause similar phenotypes: Because the alteration of different genes in a common molecular pathway can lead to similar phenotypic pathologies,22 such data can be a useful tool in scoring the proposed gene–disease relationship.20,21 Demonstration that the gene product colocalizes or physically interacts with the products of genes implicated in the same disease provides B-level evidence. When the protein colocalization and/or interaction is extended to protein families or when the phenotypic overlap between the disease in question and the known disease gene are less specific, C-level evidence is achieved.
-
Gene function: When the published literature shows that the normal function of the gene is consistent with the known biology of the disease, C-level evidence is achieved.
-
Expression profiles: Generally, mutations in disease genes cause pathology in the tissues in which the genes exhibit higher levels of expression.22 When it can be demonstrated that the expression profile of the gene of interest is strongly supportive of the proposed gene–disease relationship (such as strong expression restricted to diseased tissues), B-level evidence is achieved. When the expression profile is simply consistent with the proposed gene–disease relationship or it cannot be demonstrated that expression is restricted to diseased tissue, C-level evidence is achieved for expression, but not if a point was already counted for gene function.
Candidate-gene clinical report classification categories
-
Candidate: To reach “candidate” evidence, the alteration involved in the proposed gene–disease relationship needs to be classified as a “variant, likely pathogenic” according to Ambry’s variant classification criteria,17 which incorporate and surpass the American College of Medical Genetics and Genomics variant classification recommendations.23 Furthermore, for candidate evidence to be met, the proposed gene–disease relationship needs to score at least one point from category A, or two points from category B, or four points from category C, or one point from category B along with three points from category C.
-
Suspected candidate: In general, proposed gene–disease relationships are classified as “suspected candidates” in situations when there are sufficient points to score them as candidates but the alteration involved is classified as a variant of uncertain significance. Alternatively, proposed gene–disease relationships are classified as “suspected candidates” when candidate-level evidence is achieved but the phenotypic overlap between the evaluated patient and the previously reported patients and/or the animal models is uncertain. Last, if the alteration involved in the proposed gene–disease relationship is classified as a variant, likely pathogenic but only either one point from category B along with two points from category C or three points from category C are achieved, the proposed gene–disease relationship is scored as a suspected candidate.
-
Insufficient evidence: Alterations in genes are deemed to have insufficient supporting evidence for likely clinical relevance if any of the following apply: (i) the proposed gene–disease relationship does not meet “candidate,” “suspected candidate,” or nonreported criteria; (ii) the alteration does not affect the gene’s major isoform or the isoform is not abundantly expressed in the affected organs; (iii) the alteration is not a nonsense, frameshift, splice, or missense change at a highly conserved amino acid; or the mutant amino acid is observed as the reference in any other vertebrate species (based on species in the UCSC genome browser24); (iv) dominant inheritance is proposed for the gene–disease relationship and the alteration is observed in healthy individuals in population databases; (v) loss of function is the proposed mechanism for the gene–disease relationship among single yet heterozygous truncating alterations and other heterozygous loss-of-function alterations are observed in healthy individuals in population databases; or (vi) loss of function is the proposed mechanism for the gene–disease relationship but available data suggest functional redundancy of the gene.21
-
Nonreported: Uncharacterized gene–disease relationships are not included in patient reports in cases where the alteration does not segregate with disease in the family; is present in fewer than 30% of the reads (indicating potential mosaicism or artifact); is a single, heterozygous alteration with a minor allele frequency greater than 0.1%; or is homozygous or compound heterozygous with a minor allele frequency >0.2% in population databases.
Statistical analysis
Statistical analysis was performed using Fisher’s exact test.
Results
Rates of reported candidates and suspected candidates
Among the first 1,500 patients referred for exome sequencing, both characterized and uncharacterized genes were analyzed for 934 patients, among whom 72 (7.7%) had a reported candidate ( Table 2 ). Among these 72 patients, 37 (4.0%) received “candidate” evidence scores and 35 (3.7%) received “suspected candidate” scores.
Candidate-gene scoring
Among the 72 patients with candidate-gene reports, 71 different genes were reported. Six patients had dual findings containing two genes each ( Table 3 ). One gene (ACTG) was reported in three patients and five genes (CDC42, HNRNPK, PURA, RAD54L, and SON) were reported in each of two patients. Table 3 shows the gene, the patient’s primary reason for referral, and the candidate-gene scoring results for each patient. The references used for each evidence code are provided in Supplementary Table S1 online. The same gene could lead to a different score in different patients, as was the case with HNRNPK and SON, if (i) it was reported on different dates, since the available evidence criteria change over time, or (ii) the phenotypic overlap was uncertain for one of the patients.
DNM1 as a case example using the candidate-gene criteria
In September 2014, we reported a de novo missense alteration in the DNM1 gene in a 10-month-old boy presenting with early-onset infantile spasms, hypotonia, developmental delay, and microcephaly.
At the time, DNM1 was a clinically novel Mendelian disease gene. Analysis of the uncharacterized gene identified in the proband a heterozygous de novo alteration, DNM1 c.1190G>A (p.G397D), which was absent in his unaffected parents and dizygotic twin brother. Through application of the candidate-gene criteria scoring system, three points were awarded to the B category and two points were awarded to the C category, leading to a classification of “candidate” (Supplementary Table S2 online).
Rates of candidate-gene reports by clinical characteristics
A candidate gene was reported in patients who presented with 18 different primary reasons for referral (Supplementary Table S3 online). The highest candidate-gene report rates were among patients in the oncology/cancer susceptibility and multiple congenital anomalies categories. Age of the proband did not correlate with rates of candidate-gene findings.
Gene–disease relationships with subsequent corroborating peer-reviewed publications
Among the 71 reported candidate genes, 38% (27/71) had subsequent corroborating peer-reviewed publications describing alterations in affected patients ( Tables 1 and 4 ; Supplementary Table S4 online). Among candidate genes reaching “candidate” and “suspected candidate” evidence levels, 54.5% (18/33) and 23.7% (9/38) had subsequent corroborating publications, respectively (P = 0.014). The candidate genes described herein include those reported through November 2015. To determine whether time to publication was a factor for peer-reviewed corroboration, we further restricted the analysis to candidate genes reported through December 2014. In this cohort with at least 12 months available for subsequent corroboration (n = 52), the corroboration rate increased to 51.9% (27/52) overall and to 69.2% (18/26) among genes reaching candidate evidence and 34.6% (9/26) for genes reaching suspected candidate evidence.
Among 862 cases originally reported as negative, three (0.35%) were reclassified as positive candidates based on new publications describing patients with alterations in the gene. Prior to the publication(s), each of these genes had insufficient experimental evidence based on the candidate-gene criteria to be reported as candidates or suspected candidates.
Discussion
DES is rapidly becoming the standard of care for patients with rare diseases because it offers a one-step, unbiased interrogation of virtually all of the coding regions of the genome.
The diagnostic yield for DES among unselected patients has ranged from 25% to 40%7,15,25,26,27,28,29,30 among characterized Mendelian disease genes. In addition to our previously reported detection rate of 30% among characterized Mendelian disease genes,31 herein we report a candidate-gene rate of 7.7%, with over half of the genes (51.9%) subsequently corroborated in the peer-reviewed literature at least 12 months after clinical reporting.
Reporting candidate-gene results to patients in the context of clinical testing is important for several reasons. First, identification of the underlying genetic defect is one of the most important steps toward intervention for patients; moreover, an early and accurate diagnosis can lead to optimal care and dramatic prognostic improvements for patients and their families.7,32 New clinical management strategies have been implemented for patients receiving candidate-gene results.33 Second, as we continue to uncover additional alterations in rare disease genes, it is expected that most of these alterations will be extremely rare,4,6 and altered genes observed within single families (also known as “private genes” or “N = 1”) are expected to become more common. In this context, waiting for the identification of additional patients with an alteration in the same candidate gene may either take several years or may never occur. Third, reporting these findings to families who have been in the process of discovering their disease etiology, sometimes for several years, makes families “partners in the discovery efforts”34 and also provides patients, families, and clinicians with the opportunity to connect via social media and assist in the potential identification of additional patients with the same altered candidate gene.34 Fourth, if the discovery of new disease genes were strictly handled by research laboratories, gene discovery efforts could be hampered by problems such as lack of funding, quality standards that do not meet clinical requirements, and the possibility that patients would be barred from receiving their results.6 And last, reporting candidate-gene results to families is important because it can facilitate further research and can help connect clinicians with researchers.35
The likelihood that the identification of families with private genes (N = 1 case) will become more common also underscores the importance of having a standardized and transparent system for scoring potential candidate genes. Reliance on the availability of multiple affected families is unlikely sustainable in the near future. In such cases, the ability to make continued gene–disease discoveries must rely on other factors such as gene function and/or animal model data,6,20 as well as an overall “integrated analysis of genetic, informatic, and experimental evidence.”21 Herein, we present an integrated schema for the evaluation of candidate genes for clinical reporting. It should be noted that, as with all positive DES results, the clinician will continue to play an integral role in the ultimate interpretation of the relevance of clinical results, because the most optimal patient care comes from collaboration between the referring clinician and the diagnostic laboratory.
Further evidence for the utility of the candidate-gene scoring system to positively identify novel candidates is illustrated by the DNM1 case discussed herein. At the time of analysis, patients with pathogenic alterations in the DNM1 gene had not been reported in the literature. Based on the candidate-gene criteria, candidate-level evidence was achieved as a result of functional studies of the identical alteration in rat dynamin and the phenotype of the “fitful” mice carrying heterozygous alterations, leading to the proposal that the proband’s DNM1 alteration was causative of his epileptic encephalopathy. In less than a week after our first DNM1 clinical report, an independent peer-reviewed publication implicating de novo heterozygous DNM1 missense alterations in epileptic encephalopathies was published.36
It has been noted that candidate genes, when detected, should always be classified as of uncertain significance, requiring additional investigation and/or corroborating evidence to support their pathogenicity.23 To illustrate this, four suspected candidates were later reclassified as negative: three because of additional family studies that ruled out the alterations by failure to cosegregate with disease and one because of updated population frequency data (the Exome Aggregation Consortium database41 recently became available). Of note, none of the genes with candidate-level evidence in this cohort has been ruled out to date.
The rapid pace of novel Mendelian disease gene discoveries, made possible largely by the unique ability of clinical exome sequencing to identify patients with candidate genes, underscores the importance of having clinical validity assessment criteria that enable Mendelian disease genes to be classified as either characterized or uncharacterized. As a consequential benefit, the discovery and understanding of novel Mendelian disease genes often lead to the understanding of normal human biology and can advance our understanding of more common diseases and inspire potential diagnostic, preventive, and therapeutic opportunities20,37.
Database Information
Exome Sequencing Project: Exome Variant Server, National Heart, Lung, and Blood Institute GO Exome Sequencing Project (ESP), Seattle, WA; http://evs.gs.washington.edu/EVS/ (accessed September 2014).
Exome Aggregation Consortium, Cambridge, MA; http://exac.broadinstitute.org (accessed September 2014).
Human Gene Mutation Database; http://www.hgmd.org (accessed January 2016).
OMIM, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD; http://omim.org/ (accessed January 2016).
Orphanet: http://www.orphanet.net (accessed January 2016).
Wetterstrand KA. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP). Available at: http://www.genome.gov/sequencingcosts (accessed January 2016).
Disclosure
All the authors, with the exception of M.E.N., are employed and receive a salary from Ambry Genetics. Exome sequencing is among the commercially available tests.
References
Costa T, Scriver CR, Childs B. The effect of Mendelian disease on human health: a measurement. Am J Med Genet 1985;21:231–242.
Dodge JA, Chigladze T, Donadieu J, et al. The importance of rare diseases: from the gene to society. Arch Dis Child 2011;96:791–792.
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 2013;14:681–691.
Cooper DN, Chen JM, Ball EV, et al. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat 2010;31:631–655.
Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: 2008 update. Genome Med 2009;1:13.
Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, Soong R. Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol 2012;71:5–14.
Iglesias A, Anyane-Yeboa K, Wynn J, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med 2014;16:922–931.
Dixon-Salazar TJ, Silhavy JL, Udpa N, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 2012;4:138ra78.
Butterfield RJ, Stevenson TJ, Xing L, et al. Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurology 2014;82:1322–1330.
Rohena L, Neidich J, Truitt Cho M, et al. Mutation in SNAP25 as a novel genetic cause of epilepsy and intellectual disability. Rare Dis 2013;1:e26314.
Tuzovic L, Yu L, Zeng W, et al. A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice. Rare Dis 2013;1:e26144.
Lalani SR, Zhang J, Schaaf CP, et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am J Hum Genet 2014;95:579–583.
Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010;42:30–35.
Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet 2012;57:621–632.
Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015;17:578–586.
Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 2009;27:182–189.
LaDuca H, Stuenkel AJ, Dolinsky JS, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med 2014;16:830–837.
Tennessen JA, Bigham AW, O’Connor TD, et al.; Broad GO; Seattle GO; NHLBI Exome Sequencing Project. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012;337:64–69.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet 2012;13:565–575.
Chong JX, Buckingham KJ, Jhangiani SN, et al.; Centers for Mendelian Genomics. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet 2015;97:199–215.
MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–476.
Lage K, Hansen NT, Karlberg EO, et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA 2008;105:20870–20875.
Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Speir ML, Zweig AS, Rosenbloom KR, et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res 2016;44(D1):D717–D725.
Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014;312:1880–1887.
Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med 2012;14:51–59.
Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med 2016;18:696–704.
Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 2015;17:774–781.
Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483.
Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870–1879.
Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015;17:578–586.
Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 2011;13:255–262.
Bloss CS, Zeeland AA, Topol SE, et al. A genome sequencing program for novel undiagnosed diseases. Genet Med 2015;17:995–1001.
Might M, Wilsey M. The shifting model in clinical diagnostics: how next-generation sequencing and families are altering the way rare diseases are discovered, studied, and treated. Genet Med 2014;16:736–737.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36:928–930.
EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–370.
Green ED, Guyer MS ; National Human Genome Research Institute. Charting a course for genomic medicine from base pairs to bedside. Nature 2011;470:204–213.
MacArthur DG, Balasubramanian S, Frankish A, et al.; 1000 Genomes Project Consortium. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012;335:823–828.
Reed DR, Lawler MP, Tordoff MG. Reduced body weight is a common effect of gene knockout in mice. BMC Genet 2008;9:4.
Zaidi S, Choi M, Wakimoto H, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013;498:220–223.
Exome Aggregation Consortium; Lek M, Karczewski K, Minikel E, et al. Analysis of protein-coding genetic variation in 60,706 humans. http://biorxiv.org/content/early/2016/05/10/030338.
Acknowledgements
The authors are grateful to the patients and their families and their physicians and genetic counselors for providing samples and clinical histories.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 3095 kb)
Supplementary Table S2
(DOC 103 kb)
Supplementary Table S3
(DOC 96 kb)
Supplementary Table S4
(DOC 310 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Farwell Hagman, K., Shinde, D., Mroske, C. et al. Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet Med 19, 224–235 (2017). https://doi.org/10.1038/gim.2016.95
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.95
Keywords
This article is cited by
-
New insights into the clinical and molecular spectrum of the MADD-related neurodevelopmental disorder
Journal of Human Genetics (2024)
-
A novel heterozygous ZBTB18 missense mutation in a family with non-syndromic intellectual disability
neurogenetics (2023)
-
Application of exome sequencing for prenatal diagnosis of fetal structural anomalies: clinical experience and lessons learned from a cohort of 1618 fetuses
Genome Medicine (2022)
-
Homozygous variant in MADD, encoding a Rab guanine nucleotide exchange factor, results in pleiotropic effects and a multisystemic disorder
European Journal of Human Genetics (2021)
-
Neurofascin antibodies in chronic inflammatory demyelinating polyradiculoneuropathy: from intrinsic genetic background to clinical manifestations
Neurological Sciences (2021)
