
Abstract
Purpose:
Germ-line mutations in the exonuclease domains of POLE and POLD1 have been recently associated with polyposis and colorectal cancer (CRC) predisposition. Here, we aimed to gain a better understanding of the phenotypic characteristics of this syndrome to establish specific criteria for POLE and POLD1 mutation screening and to help define the clinical management of mutation carriers.
Methods:
The exonuclease domains of POLE and POLD1 were studied in 529 kindred, 441 with familial nonpolyposis CRC and 88 with polyposis, by using pooled DNA amplification and massively parallel sequencing.
Results:
Seven novel or rare genetic variants were identified. In addition to the POLE p.L424V recurrent mutation in a patient with polyposis, CRC and oligodendroglioma, six novel or rare POLD1 variants (four of them, p.D316H, p.D316G, p.R409W, and p.L474P, with strong evidence for pathogenicity) were identified in nonpolyposis CRC families. Phenotypic data from these and previously reported POLE/POLD1 carriers point to an associated phenotype characterized by attenuated or oligo-adenomatous colorectal polyposis, CRC, and probably brain tumors. In addition, POLD1 mutations predispose to endometrial and breast tumors.
Conclusion:
Our results widen the phenotypic spectrum of the POLE/POLD1-associated syndrome and identify novel pathogenic variants. We propose guidelines for genetic testing and surveillance recommendations.
Genet Med 18 4, 325–332.
Similar content being viewed by others

Introduction
Germ-line mutations in the exonuclease (proofreading) domain of DNA polymerases Pol δ and Pol ɛ have been associated with a dominantly inherited syndrome that confers increased risk to colorectal cancer (CRC) and polyposis.1 Two recurrent pathogenic variants, POLE p.L424V and POLD1 p.S478N, have been identified in 21 and 3 families, respectively.1,2,3,4 A novel mutation in POLD1, p.L474P, was found in a hereditary nonpolyposis CRC family.2
Patients carrying POLE and POLD1 exonuclease domain mutations show variable phenotypes including multiple adenomas and CRC, and endometrial cancer in the case of female POLD1 mutation carriers.1,2,3,4 A better characterization of the syndrome is currently required to establish specific criteria for POLE and POLD1 exonuclease mutation screening and to help define the clinical management of mutation carriers. To fulfill this aim, here we study the complete exonuclease domains of POLE and POLD1 in 529 independent families characterized by the presence of familial or early-onset mismatch repair (MMR), proficient CRC, and/or APC-negative and MUTYH-negative polyposes.
Materials and Methods
Study sample
A total of 544 CRC cases belonging to 529 families were included in the study: 456 familial CRC cases from 441 uncharacterized MMR-proficient families, including 60 Amsterdam-positive families, and 88 polyposis cases. Most of them, 526 cases (511 families), were previously genotyped for POLE p.L424V and POLD1 p.S478N (included in series no. 1 in the work by Valle et al.2). All of them were referred to the Genetic Counseling Units of the Catalan Institute of Oncology in the Spanish region of Catalonia between 1999 and 2012. Referral was based on family history of CRC or polyps, presence of early-onset CRC, and/or personal history of polyposis. Eighteen additional CRC patients belonging to unrelated Amsterdam I MMR-proficient families were included in the study. These were recruited through the Human Genetics Program of the Spanish National Cancer Research Center (CNIO).
Among the 456 MMR-proficient cases (441 families), 49 (10.7%) fulfilled Amsterdam I, 11 (2.4%) fulfilled Amsterdam II, and 390 (85.5%) fulfilled the Bethesda criteria. No specific information on family history was available for six patients. The mean age at cancer diagnosis was 48.98 (SD: 12.54) for the tested individuals. Nonpolyposis cases were MMR-proficient, i.e., their tumors showed microsatellite stability and expression of the MMR proteins MLH1, MSH2, MSH6, and PMS2.
Clinical features of polyposis cases, which included adenomatous polyposes (17.0%), attenuated adenomatous polyposes (47.7%), and nonadenomatous polyposes (15.9%), were detailed by Valle et al.2 For these cases, screening of MUTYH and APC mutations was performed as previously described.2
Informed consent was obtained from all subjects and the study received the approval of the Ethics Committee of the Institut d'Investigació Biomèdica de Bellvitge (IDIBELL) (PR073/12).
Germ-line mutation identification in pooled samples
Mutation screening of the exonuclease coding regions of POLE and POLD1 was performed by using a combination of pooled samples, PCR amplification (POLE exons 9–14 and POLD1 exons 6–12), and high-throughput sequencing, as previously described.5 Amplification of the DNA pools was performed using Phusion High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA) and custom-designed primers covering the exons and intron–exon boundaries (Supplementary Table S1 online). Next-generation sequencing was performed on a HiSeq-2000 at the Centro Nacional de Análisis Genómico (CNAG, Barcelona, Spain).
Direct automated sequencing
Direct automated (Sanger) sequencing was used to validate the results obtained by massive parallel sequencing, to identify the mutated cases within pools, and to perform the co-segregation studies. Sequencing was performed on an ABI Sequencer 3730 (Applied Biosystems, Life Technologies, Foster City, CA) using a standard protocol. Data were analyzed using Mutation Surveyor (version 3.10) (Softgenetics, State College, PA, USA). The primers used were the same as those used for germ-line mutation identification in pooled samples (Supplementary Table S1 online).
In silico prediction analysis
Protein damage prediction of missense genetic variants was performed by using the in silico algorithms PolyPhen-2, SIFT, Condel, and Mutation Taster.6,7,8,9 dbNSFP (version 2.8) was used to obtain the evolutionary conservation PhyloP scores.10 Possible alterations of the splice sites were evaluated using NNSplice0.9.11
Structural analysis
The human 3D model of POLD1 was obtained from ModBase (http://modbase.compbio.ucsf.edu/) and improved by the RepairPDB and Optimize commands of FoldX (http://foldx.crg.es). This model was calculated using as a template the crystallographic structure of the homologous yeast protein Pol3 (PDB ID: 3iay, chain A), which shares 51% sequence identity with the human POLD1.12 Based on this model and domain annotations from the yeast protein Pol3, the exonuclease domain of POLD1 comprises amino acids R311-L526. For the identified variants, protein stability calculations were performed using CUPSAT (http://cupsat.tu-bs.de), I-Mutant 2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0.html), ERIS (http://troll.med.unc.edu/eris/), and PoPMuSiC (http://dezyme.com).
Loss of heterozygosity
Six microsatellite flanking POLD1 and spanning 2.06 Mb, three centromeric (D19S867, D19S585, D19S904) and three telomeric (D19S246, D19S907, and D19S601), were used to assess loss of heterozygosity (LOH) in DNA extracted from formalin-fixed paraffin-embedded tissue. Also, SNaPshot targeting the corresponding POLD1 mutation was used to assess LOH and to discriminate wild-type and mutated alleles. LOH was scored if the intensity of any allele was reduced by ≥50% relative to the other allele after taking account of the relative allelic intensities in paired constitutional DNA. Primers and conditions are shown in Supplementary Table S1 online.
Results and Discussion
Germ-line mutations in the exonuclease domain of POLE and POLD1 in a series of familial CRC and/or polyposis
In a previous study, we analyzed by targeted genotyping the recurrent mutations POLE p.L424V and POLD1 p.S478N in a Spanish familial CRC series, which included 438 MMR-proficient familial nonpolyposis (423 families) and 88 unrelated polyposis CRC patients, among others (series no. 1).2 We identified a de novo POLE p.L424V in a CRC and polyposis patient with anaplastic oligodendroglioma recently diagnosed at age 30.
Here, we extended the analysis by sequencing the complete exonuclease domains of POLE and POLD1 in the aforementioned patients, adding 18 additional MMR-proficient Amsterdam I–positive families, thus making a total of 456 MMR-proficient familial nonpolyposis CRC cases (441 families) and 88 polyposes. It corresponds to the largest series of hereditary nonpolyposis CRC where the complete exonuclease domains of POLE and POLD1 have been studied.
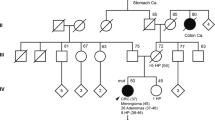
Although no additional mutations were identified in POLE, six novel or rare nonsynonymous variants were detected in POLD1, five of them in previously genotyped patients. In silico predictions, evolutionary conservation, absence in control populations, functional effects on yeast,13,14 and/or affectation of an exonuclease catalytic residue, strongly support the pathogenic effect of four of the six identified variants: POLD1 p.D316H, p.D316G, p.R409W, and p.L474P ( Table 1 ). Localization within the exonuclease domain of the amino acids affected by these mutations is shown in Supplementary Figure S1 online. CRC was diagnosed in 62.5% (5/8) of mutation carriers, and both endometrial and breast cancers were diagnosed in 33.3% (2/6) of female carriers; one was diagnosed with two primary breast tumors (see complete tumor spectrum of mutated families in Figure 1 and Supplementary Table S2 online). Only one POLD1 mutation carrier (1/8; 12.5%) was diagnosed with more than five colonic adenomas (cumulative resection of 11 adenomas until age 67), suggesting that POLD1 mutations do not show strong association with polyposis and agreeing with the findings observed in mice.15

Pedigrees of the families with germ-line POLD1 variants. Demonstrated pathogenic variants are indicated in bold, and variants of unknown significance are shown in regular case. Filled symbol, cancer; centered gray circle, presence of ≥2 polyps (adenomatous or hyperplastic); +, mutation carrier; −, wild-type. Ages at information gathering or at death (†), when available, are indicated on the top-left corner. Probands are depicted by an arrow. End, endometrial cancer; Br, breast cancer; Lymph, lymphoma.
Not so evident are the functional effects of POLD1 p.V295M and p.R521Q (minor allele frequencies ≤0.035%), which are classified as variants of unknown significance ( Table 1 ). However, p.V295M, outside but close to the exonuclease domain (Supplementary Figure S1 online), occurs in two independent affected families ( Figure 1 ). In one of them, p.V295M is found in trans with p.D316G (catalytic residue) (individual Fam-2, II.2). The patient was diagnosed with endometrial cancer and two metachronous breast tumors. To date, no biallelic mutation in POLE or POLD1 has been reported. Considering the likely pathogenic variants identified in this study, POLD1 exonuclease mutations account for 1% (4/411) of MMR-proficient familial/early-onset nonpolyposis CRC cases, including 1.7% (1/60) of Amsterdam-positive cases (familial CRC type X).
Review of the phenotypic data from the reported carriers of POLE and POLD1 exonuclease pathogenic mutations
Phenotypic data from the 69 carriers (29 families) of POLE/POLD1 exonuclease pathogenic mutations reported to date (refs. 1,2,3,4 and in the present study) are recapitulated in Table 2 (POLE) and Table 3 (POLD1) and are summarized according to the mutated gene in Table 4 . Altogether, available information point to an associated phenotype characterized by attenuated or oligo-adenomatous colorectal polyposis (>80% of POLE and >60% of POLD1 mutation carriers were diagnosed with ≥2 adenomas; on average, 16 adenomas), CRC (60–64% of carriers), and probably brain tumors (5.8%). Gastroduodenal (mostly duodenal) adenomas were detected in 57.1% of carriers who underwent gastroduodenoscopies (only 14 POLE exonuclease mutation carriers evaluated).4 Moreover, the POLD1 phenotypic spectrum includes endometrial (57.1% of female carriers) and breast tumors (14.3% of female carriers). All 21 POLE/POLD1 mutation carriers without cancer underwent resection of colorectal adenomas, indicating complete or very high expressivity of the associated phenotype (associated carcinomas and/or adenomas), but precluding any conclusion about CRC penetrance.
Ascertainment bias due to the inclusion of CRC and/or polyposis families in the studies may have led to overrepresentation or under-representation of the POLE/POLD1–associated tumors. In particular, the high prevalence of endometrial cancer in POLD1 families might be biased by the inclusion of families fulfilling the Amsterdam or Bethesda criteria. However, the fact that endometrial cancer is extremely rare among female POLE mutation carriers (1/26 compared to 8/14 female POLD1 carriers) supports its relevance in the POLD1-associated phenotype.
Recommendations for genetic testing and clinical surveillance
Clinical suspicion of the polymerase proofreading-associated syndrome may arise when the clinical characteristics depicted in Table 4 (left column) are fulfilled. Due to the current need of genetic testing guidelines for POLE/POLD1, we attempt to define the first preliminary recommendations for their use in the routine practice. Based on the overlapping phenotypes of this entity with Lynch syndrome and attenuated adenomatous polyposis (APC/MUTYH),16 we took as a model the criteria for hereditary nonpolyposis CRC (revised Bethesda), adapted them to the attenuated or oligo-polyposis scenario, and took into consideration additional specific POLE/POLD1 characteristics. The resulting recommendations are shown in Table 4 (right column). Although preliminary, they will help guide in routine genetic testing and counseling until larger series of mutation carriers are described and standardized guidelines defined.
The implementation in routine genetic testing of targeted next-generation sequencing using multi-gene panels will help alleviate the issue of overlapping phenotypes among familial CRC and polyposis syndromes. In this context, the proposed guidelines may be used for prioritization purposes in the analysis of data.
More extensive phenotypic data from mutation carriers are needed to establish standardized surveillance recommendations. In the meantime, based on the clinical features of POLE and POLD1 mutation carriers ( Table 4 ) and the guidelines recommended for Lynch syndrome and attenuated adenomatous polyposis,17 whose features largely overlap with those observed in the polymerase proofreading-associated syndrome, we recommend colonoscopies every 1–2 years and gastroduodenoscopies every 3 years, starting at age 20–25 (reevaluate periodicity according to the findings), adding endometrial cancer screening beginning at age 40 for POLD1 female carriers. The predisposition to breast tumors may eventually influence the age to start and the frequency of mammogram screenings. For both POLE and POLD1 mutation carriers, the possibly increased susceptibility to brain tumors may be taken into consideration.
Disclosure
The authors declare no conflict of interest.
References
Palles C, Cazier JB, Howarth KM, et al.; CORGI Consortium; WGS500 Consortium. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013;45:136–144.
Valle L, Hernández-Illán E, Bellido F, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet 2014;23:3506–3512.
Elsayed FA, Kets CM, Ruano D, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet; e-pub ahead of print 5 November 2014.
Spier I, Holzapfel S, Altmüller J, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer; 2015; 15;137(2):320–31.
Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011;475:101–105.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081.
González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 2011;88:440–449.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–362.
Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 2013;34:E2393–E2402.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol 1997;4:311–323.
Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol 2009;16:979–986.
Kokoska RJ, Stefanovic L, DeMai J, Petes TD. Increased rates of genomic deletions generated by mutations in the yeast gene encoding DNA polymerase delta or by decreases in the cellular levels of DNA polymerase delta. Mol Cell Biol 2000;20:7490–7504.
Murphy K, Darmawan H, Schultz A, Fidalgo da Silva E, Reha-Krantz LJ. A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome 2006;49:403–410.
Albertson TM, Ogawa M, Bugni JM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci USA 2009;106:17101–17104.
Hampel H, Bennett RL, Buchanan A, Pearlman R, Wiesner GL ; Guideline Development Group, American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 2015;17:70–87.
Stoffel EM, Mangu PB, Gruber SB, et al.; American Society of Clinical Oncology; European Society of Clinical Oncology. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol 2015;33:209–217.
Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2012;44:47–52.
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (State Secretariat for Research, Development, and Innovation; SAF2012-38885 and SAF2010-21165, Ramón y Cajal contract to L.V., fellowship to F.B., and RTICC (Red Temática de Investigación Cooperativa en Cáncer) networks RD12/0036/0031, RD12/0036/0008, and RD12/0036/0067), the Scientific Foundation Asociación Española Contra el Cáncer, the Government of Catalonia (2014SGR-338), and the EU FP7 project ASSET (grant agreement 259348 to A.V.).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figures and Tables
(DOC 1196 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Bellido, F., Pineda, M., Aiza, G. et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med 18, 325–332 (2016). https://doi.org/10.1038/gim.2015.75
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.75
Keywords
This article is cited by
-
Discovery of recessive effect of human polymerase δ proofreading deficiency through mutational analysis of POLD1-mutated normal and cancer cells
European Journal of Human Genetics (2024)
-
Multiple duodenal epithelial tumors in a patient with polymerase proofreading-associated polyposis in POLE variant
Clinical Journal of Gastroenterology (2024)
-
Solving the enigma of POLD1 p.V295M as a potential cause of increased cancer risk
European Journal of Human Genetics (2022)
-
The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management
Familial Cancer (2022)
-
Unravelling roles of error-prone DNA polymerases in shaping cancer genomes
Oncogene (2021)
