
Abstract
Purpose:
To understand the translational trajectory of genomic tests in cancer screening, diagnosis, prognosis, and treatment, we reviewed tests that have been assessed by recommendation and guideline developers.
Methods:
For each test, we marked translational milestones by determining when the genomic association with cancer was first discovered and studied in patients, and when a health application for a specified clinical use was successfully demonstrated and approved or cleared by the US Food and Drug Administration. To identify recommendations and guidelines, we reviewed the websites of cancer, genomic, and general guideline developers and professional organizations. We searched the in vitro diagnostics database of the US Food and Drug Administration for information, and we searched PubMed for translational milestones. Milestones were examined against type of recommendation, Food and Drug Administration approval or clearance, disease rarity, and test purpose.
Results:
Of the 45 tests we identified, 9 received strong recommendations for their usage in clinical settings, 14 received positive but moderate recommendations, and 22 were not currently recommended. For 18 tests, two or more different sources had issued recommendations, with 67% concordance. Only five tests had Food and Drug Administration approval, and an additional five had clearance. The median time from discovery to recommendation statement was 14.7 years.
Conclusion:
In general, there were no associations found between translational trajectory and recommendation category.
Genet Med 17 6, 431–440.
Similar content being viewed by others

Introduction
The promise of genomics is expected to lead to the development of precision or personalized tests that can be used in disease diagnosis, treatment, and prevention.1 However, much of our understanding of the genomic component of disease is still limited, and the appropriateness of genomic tests in various clinical settings is even more uncertain.2 The apparent inconsistency in regulation of genomic tests further exacerbates the prevailing ambiguity. The US Food and Drug Administration (FDA) regulates some tests but exercises “enforcement discretion” when it comes to oversight of laboratory-developed tests.3 Neither regulation approach necessarily takes into consideration the clinical utility of a test.
Professional organizations and guideline developers try to fill in the regulatory gaps and provide guidance to stakeholders on genomic tests by evaluating the tests and commenting on their clinical appropriateness. Nevertheless, differential evaluative methods used across these organizations can result in added confusion instead of providing the needed clarity. For example, recommendations can be based on ratings of analytical validity, clinical validity, and clinical utility,4 and/or contextual factors such as cost and psychosocial considerations.4,5 Some organizations require rigorous systematic reviews to generate evidence-based guidelines/recommendations,4,5,6,7,8 whereas others rate the consensus of expert opinions.6,9
Recommendations and guidelines are at the end of a long process of research and development that includes several intermediate translational milestones. These include the discovery of the potential association between a genetic marker and disease, its study of patients with the disease of interest, and the successful clinical demonstration of the health application. There is no systematic evaluation to date regarding how long it takes for these milestones to be accomplished for genomic tests.
Here, we systematically review genomic tests in which recommendations for or against their use in clinical practice have been made in the screening, diagnosis, prognosis, or treatment of diverse types of malignancies. We examine how recommendations agreed or disagreed across recommendation sources, which may have particular implications in the clinical scenario when providers are faced with conflicting recommendations. By determining how various milestones and testing characteristics were met and how recommendations were made, we may better understand the translational trajectory of genomic tests and may gain some insights on improving the efficiency of the translational process.
Materials and Methods
Identifying recommendations
Genomic tests were selected by first identifying recommendations issued on the application of genomic testing for cancer-related purposes. These include recommendation statements and clinical practice guidelines made by professional organizations and guideline development groups on testing for single genes and multigene testing products. Specifically, we reviewed the websites for the National Comprehensive Cancer Network (NCCN),9 the American Society for Clinical Oncology (ASCO),6 the Evaluation of Genomic Tests in Practice and Prevention (EGAPP) Working Group,4 the United States Preventive Services Task Force,8 the Blue Cross Blue Shield Technology Evaluation Center,7 the National Institute for Health and Clinical Excellence,5 and the National Guideline Clearinghouse.10 Although most source websites list only self-published guidelines, National Guideline Clearinghouse includes clinical practice guidelines published by numerous organizations, identified through searches of participating guideline developer websites, literature searches, and user submissions. Potential recommendations were identified using the “neoplasm” filter on the National Guideline Clearinghouse database. Finally, we reviewed the NCCN publication by Febbo et al.11 to identify additional tests.
From each recommendation, we abstracted the following information: the gene(s) on which the test was based; the associated cancer type(s); the date the assessment was published; earlier recommendations, if available; and strength of evidence and/or strength of recommendation when available.
We assigned tests to one of three recommendation categories based on information provided by the recommendation sources:
-
Category 1: Tests with strong support for use of the test in clinical practice. These tests were recommended based on high-level evidence and/or had strong wording advocating their use (e.g., “should be used”).
-
Category 2: Tests that had some support for use in clinical practice, but recommendation statements mention that the statement is based on lower-level evidence and/or the need for more robust evidence.
-
Category 3: Tests that did not have support for current use in routine clinical practice. These included tests for which statements mentioned insufficient evidence for encouraging or discouraging current use.
In some instances, sources used rating systems to describe the strength of their recommendation and/or evidence supporting their recommendation (e.g., NCCN uses a rating system in which their category 1 indicates that “based on high-level evidence, there is uniform NCCN consensus that the intervention is appropriate”). Other sources relied on specific language to describe the nature of their recommendation (e.g., EGAPP deems tests as either those with sufficient evidence or convincing evidence or those with insufficient evidence). We used all available information provided by sources to inform categorization of each test (Supplementary Table S1 online). We considered categories 1 and 2 as positive recommendations, and category 3 as a negative recommendation.
There were some cases of disagreement in recommendations between sources commenting on the same application. To categorize the recommendation, we used the majority recommendation (e.g., if three sources did not recommend use of testing but one source did, then we deferred to the majority recommendation and categorized the test as category 3) or, in cases in which there were equal numbers of disagreeing recommendations, the more favorable recommendation was used for analysis.
For tests with multiple applications (i.e., used for predicting recurrence and predicting response to treatment), we also assigned recommendation categories specific for each test purpose. We used these categories when analyzing tests by test purpose. For the overall analysis of tests with multiple applications, the most favorable recommendation was used.
Inclusion/exclusion criteria
Only those tests that assess DNA, RNA, or protein expression qualified as a genomic or genetic test. We included tests that related to cancer screening, diagnosis, prognosis, or drug treatment. All types of cancer were eligible, but we excluded cancer precursors.
Determining test FDA approval or clearance
Each gene identified was searched in the in vitro diagnostics database of the FDA website.12 Search results were examined and the date of the earliest approved or cleared test was noted. FDA approval is given to medical devices for which the applicant provides reasonable assurance of the device’s safety and effectiveness in a premarket approval application. FDA clearance is given to medical devices for which the applicant shows that the medical device is “substantially equivalent” to another already legally marketed for the same use using the 510(k) pathway.12 For a test to be considered approved or cleared in our study, it must have been approved or cleared for the same cancer and use for which the recommendation was made. Tests for which we found no search results were noted as neither approved nor cleared.
Identifying translational milestone dates
We identified landmark events in the translational life cycle of a genomic test to track progress and timing. We followed an approach that has been applied previously for evaluating translational milestones,13,14 and we tailored it to the types of tests that we examined in this overview. Three milestones were considered:
-
1
When the association between the specified cancer and gene was first identified. For example, if a test is specific for thyroid cancer, we used the first study that identified the association between that gene and thyroid cancer. In some cases, discovery was through animal or cell studies. For multigene assays, we used the date of the first publication on assay development.
-
2
The first human study of the cancer–gene association. We only considered studies that used patient samples (i.e., excluded cell lines).
-
3
The first successful clinical demonstration of the test in which patients underwent testing and the results were informative for the purpose(s) specified by the recommendation. For tests that had multiple purposes (e.g., prognostic and pharmacogenomic), we obtained dates for the earliest studies demonstrating each purpose. We considered a test to have successfully demonstrated a clinical application if the study’s authors concluded so in the abstract.
To collect milestone information, we searched PubMed for publications using a simple query of the respective gene(s) and cancer type(s). Genes with historically different names or symbols not approved by the Human Genome Organization were also searched using the alternate names. We excluded non-English-language publications. We reviewed the abstracts starting from the earliest publication date, and the full-text of the earliest relevant article was read to ascertain relevance. If the article referenced earlier studies on the topic of interest, we examined those studies and determined relevance. When it was evident that an article was the first instance in which that milestone was documented, we noted the publication date. If the day or month of publication was not given, then the earliest possible day or month was entered.
Two independent reviewers (C.Q.C. and S.R.T.) conducted the literature review. Approximately half of the tests were redundantly searched by the reviewers and, because of agreement between the results, it was determined that there was consensus in the review methods. All searches were last updated on 30 October 2013.
Analyses
We presented descriptive information on the identified eligible tests, including gene(s) involved, type(s) of cancer, test purpose (screening, diagnosis, prognosis, or treatment), the source and nature of relevant recommendations, whether recommendations were positive or negative and the respective category (1, 2, or 3), how many sources made recommendations, whether recommendations from different sources agreed, and whether there was FDA approval, clearance, or neither.
We compared tests with different types of recommendations and with or without FDA approval in terms of type of cancer (rare or common), type of test (screening, diagnostic, prognostic, or pharmacogenomic), and the timing of their translational milestones. In accordance with the National Cancer Institute, we labeled tests as rare if their specified cancer incidence was <40,000 cases in 2013.15 In SPSS (IBM SPSS Statistics for Windows, version 22.0; SPSS, Armonk, NY), we used exact tests for 2 × 2 and 2 × 3 tables, and we used Mann–Whitney U-test and Kruskal–Wallis test for comparisons of continuous measures in two or three groups, respectively. Kaplan–Meier plots were drawn to show the time from first discovery to the first issuance of a recommendation statement of the same type as the final recommendation (i.e., first positive recommendation for a test having a positive final recommendation) for each category of tests. All P values provided are two-sided, with significance at P < 0.05. Genomic tests were also stratified by multigene (n = 5) and single gene tests (n = 40) for comparison with translational trajectory and recommendation category.
Results
Figure 1 illustrates the identification process and exclusion criteria. We screened 941 recommendations and guidelines and identified 45 unique cancer genomic tests.

Search strategy and selection of cancer genomic tests.
Description of tests
Table 1 presents selected characteristics of the 45 eligible tests. Most tests were for use in colon/colorectal cancer (n = 11) and acute myeloid leukemia (n = 11), followed by non–small cell lung cancer (NSCLC) (n = 6), glioma (n = 5), and breast cancer (n = 4). Acute lymphocytic leukemia, chronic lymphocytic leukemia, breast and ovarian cancers, thyroid cancer, melanoma, and prostate cancer each had one test. Two tests were directed for pharmacogenomic use in multiple malignancies. Overall, 19 tests were for rare cancers. Twenty-seven tests were intended for prognostic use, 23 were intended for pharmacogenomic use, 6 were intended for diagnostic use, and 4 were intended for screening use. Eleven tests had two or more uses and are thus represented in more than one test use category.
Recommendations and FDA status
Table 1 shows the distribution of recommendations made by professional organizations. The NCCN commented on 71% (n = 32) of tests, followed by EGAPP (n = 9) and ASCO (n = 9). Based on our categorizations, 23 (51%) of the eligible genomic tests received positive recommendations, with 9 category 1 (strong recommendation/high-level evidence) and 14 category 2 (favorable recommendation/lower-level evidence) ( Table 1 ).
Twenty-two percent of genomic tests received either FDA approval (n = 5) or clearance (n = 5) ( Table 1 ). As shown, most of the tests with category 1 recommendations had FDA approval or clearance (5/9); however, this was uncommon for category 2 (1/14) and category 3 (4/22) recommendations (P = 0.017). Four tests with category 1 recommendations (screening for Lynch syndrome, 1p/19q for glioma, oncotype DX for breast cancer, and RET for thyroid cancer) have not received FDA approval or clearance. By contrast, one test with a category 3 recommendation has FDA approval (PCA3 for prostate cancer), and three have FDA clearance (CYP2D6 for breast cancer, Mammaprint for breast cancer, and TP53 for colorectal cancer). One category 2 test received FDA clearance (UGT1A1 for colorectal cancer).
Eighteen genomic tests received recommendation statements from two or more organizations. There was an average of two recommendations per test. Seven tests received recommendation statements from more than two organizations. Because 12 of the 18 tests had unanimous agreement across recommendations, the concordance of recommendations was 67%. We observed differing recommendations for six genomic tests (BRAF, CYP2D6, ERCC1, Oncotype DX, RRM1, and UGT1A1) (Supplementary Table S2 online). Discrepancies in recommendations may be attributable to emerging evidence favoring use of these tests and/or differences in evidence appraisal.
We observed changes in recommendation within a single organization for three genomic tests (ERCC1, EGFR, and KRAS). For ERCC1, the NCCN cited insufficient evidence in 2011 but gave it a positive recommendation 2 years later. In 2007, the Blue Cross Blue Shield Technology Evaluation Center determined that there was insufficient evidence for EGFR mutation testing to guide treatment; later, in 2011, it issued a positive recommendation for the same application. Similarly, the NCCN task force report in 2011 (ref. 11) and the NCCN Guidelines for NSCLC at the time did not recommend KRAS testing for prognostic or predictive purposes, but their most recent guideline in 2013 states that testing could be useful for selection of candidates for tyrosine kinase inhibitor therapy.9 These changes were likely attributable to emerging evidence favoring use of these tests.
Translational milestones
Supplementary Table S3 online shows the translational milestones for the 45 tests. The median time from discovery to first human study was 0 (interquartile range (IQR): 0–0) years, the median time from first human study to first demonstration of test was 3.9 (IQR: 1.7–8.2) years, and the median time from first demonstration of test to earliest recommendation issued of the same type as the final recommendation was 6.1 (IQR: 1.9–11.3) years. The median time from discovery to earliest recommendation issued of the same type as the final recommendation was 14.7 (IQR: 6.7–19.7) years.
When limited to the 10 tests that had FDA approval or clearance, the median time from discovery to first human study was 0 (IQR: 0–0.3) years, the median time from first human study to first demonstration of test was 5.6 (IQR: 2.5–16.0) years, and the median time from first demonstration of test to FDA approval or clearance was 5.4 (IQR: 0.9–11.8) years. The median time from discovery to FDA approval was 13.6 (IQR: 7.1–26.6) years.
When limited to the 23 tests with positive recommendations, the median time from discovery to first human study was 0 (IQR: 0–1.2) years, the median time from first human study to first demonstration of test was 3.9 (IQR: 2.4–9.9) years, and the median time from first demonstration of test to first recommendation was 7.1 (IQR: 2.9–11.7) years. The median time from discovery to first recommendation statement issued was 14.8 (IQR: 9.3–22.1) years, and from discovery to the first recommendation matching the same type as the most recent recommendation, the median time was 14.9 (IQR: 11.3–22.1) years.
Correlates of types of recommendations
As shown in Table 2 , there was no association between tests for rare cancers and recommendation category, although they were less likely to have FDA approval or clearance (P = 0.029). Analyzing by test purpose (e.g., tests that had pharmacogenomics uses associated with it were analyzed together), we found no relationship between test purpose and recommendation category. However, tests intended for prognostic purposes were less likely to have FDA approval or clearance (P = 0.008).
The distribution of all three milestone dates did not significantly vary across recommendation categories. Observations were similar when we compared positive recommendations with category 3 recommendations. For the 10 FDA-approved or -cleared tests, the dates of approval or clearance were not associated with recommendation category.
We also assessed whether the time between translational milestones and other events influenced recommendation. The overall analyses and subgroup analysis by test purpose indicated that the amount of time from discovery to test demonstration did not vary across recommendation groups or FDA status. However, a long recommendation history (defined as the amount of time between the most recent recommendation statement to the earliest recommendation statement with the same recommendation type; i.e., if the most recent recommendation was positive, then we used the earliest positive recommendation found) was associated with recommendation category. In comparing medians across recommendation categories, the length of recommendation history was 2.3 years longer for category 1 tests compared with category 2 tests and was 1.6 years longer for category 2 tests compared with category 3 tests (P = 0.010). Findings were similar when comparing FDA-approved tests with unapproved tests; approved tests had a longer recommendation history by 1.4 years (P = 0.039).
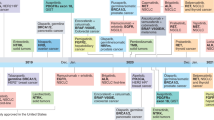
Figure 2 summarizes the trajectory paths of all 45 tests, from discovery to the earliest recommendation matching the final recommendation type, across recommendation categories. The median number of years in the life of a genomic test for category 1, 2, and 3 tests were 15.7, 14.8, and 11.8 years, respectively. These differences were not statistically significant (P = 0.308).

Time from discovery to earliest recommendationa for cancer genomic tests, by recommendation category. aEarliest recommendation of the same type as the most recent recommendation.
Stratification by test type
Results were similar when stratifying by multigene and single gene tests. There was no association between translational trajectory and recommendation category (multigene tests P = 1.00; single-gene tests P = 0.616).
Discussion
Our review identified 45 cancer genomic tests from 15 professional organizations or guideline developers. Nearly half were positively recommended, but only a minority was strongly recommended for use in clinical practice. We observed a number of tests that had received different recommendations, over time and/or by different organizations. FDA approval or clearance was seen mostly in tests with strong recommendations, but the correspondence between FDA and other organizations’ recommendations was only modest. The examined tests took a long time (on average 15 years) to reach from their initial discovery to the point of being discussed in recommendations, and the time was equally long regardless of the type of recommendation.
Genomic tests with the strongest recommendations tended to have been in existence the longest, and thus may benefit from accumulated evidence reflected by a longer recommendation history in comparison with other tests with lower levels of recommendation. Likewise, moderately recommended tests tended to have a longer history than those not recommended. One explanation for this pattern may be that as the evidence base develops and increases in quality, an organization may change a previously moderate recommendation to a strong recommendation, which requires a high level of evidence. Moderate recommendations are generally based on emerging or low-level evidence. By contrast, most negative recommendations had little or conflicting evidence for their use because of the short history. Therefore, organizations are unlikely to comment on the thousands of genetic markers proposed by initial studies until there is more concrete evidence. We observed a similar correlation between recommendation history and categories of recommendation when we classified the tests by FDA status.
We found that tests for rare cancers were less likely to have FDA approval or clearance than tests for common cancers. This may be because it is easier to conduct studies for common diseases than for those afflicting a small population. Our analysis also revealed that tests intended for prognostic purposes were less likely to have FDA clearance than other tests. Others have observed that compared with other research designs, prognostic studies are ubiquitous, but the questionable quality and mostly retrospective nature of the data have yielded few clinically useful markers.16,17 Nevertheless, neither cancer rarity nor prognostic use was associated with recommendation type.
We observed that there was a significant association between FDA status and recommendation category. FDA approval was the influential factor for the association, because there was no detected difference when comparing clearance with recommendation. This is unsurprising because FDA-approved companion diagnostics are required for the use of specific drugs. However, we also observed several discrepancies when FDA approved/cleared status was not commensurate with otherwise negative recommendations by other organizations.
Our analysis shows that a strongly recommended genomic test takes nearly 16 years from discovery to recommendation. Our finding corroborated that of previous publications indicating that there is a 17-year translational period between scientific discovery and clinical practice.18 Perhaps it is not surprising that the path to clinically actionable tests is muddy and convoluted when examining biomarkers within the larger contextual landscape. It is estimated that more than 150,000 articles have been published on 1,000 claimed biomarkers, but less than 10% of these biomarkers have been validated for routine clinical practice.19 Given that genetic markers are a subset of biomarkers, the number of validated genetic tests is therefore likely to be vastly smaller, and thus the translational road would be at least as long and convoluted.
Our analysis showed no correlation between translational trajectories of cancer genomic tests and recommendation categories. This suggests that the translational trajectories are unpredictable and heterogeneous within recommendation categories. Reasons for this observation may vary and may be specific for each test. For instance, both BRAF testing for vemurafenib use and TPMT testing for mercaptopurine use are examples of category 1 pharmacogenomic tests, but BRAF has a short trajectory and TPMT has a long trajectory. This difference may be that the BRAF and vemurafenib were discovered at the same time,20 whereas mercaptopurine was in use long before its connection to TPMT was made.21,22 Another is that, although both are considered pharmacogenomic, BRAF testing is used to select melanoma patients who will respond to vemurafenib treatment, whereas TPMT testing is used for dose adjustment of mercaptopurine.12
We included several multigene assays, which have a different trajectory starting point than single-gene tests, primarily because they are developed specifically for clinical use and start with the patients. The time between milestones is often shorter for these assays. For example, the time between discovery and first recommendation for Mammaprint, a 70-gene signature used to stratify risk of breast cancer recurrence,23 was 5 years. It was discovered by microarray analysis of primary breast tumors in 2002, received FDA clearance in 2007, and first recommendation was noted in 2009. Stratification by multigene assay (Coloprint, H:I Ratio, Oncotype DX Breast, Oncotype DX Colon, Mammaprint) and single-gene test type showed no association between translational trajectory and recommendation category (multigene tests P = 1.00; single-gene tests P = 0.616).
Among the 18 tests that were recommended by multiple organizations, we observed good, but not excellent, concordance in recommendations. In the cases in which opposing recommendations were made, we speculated possible reasons for the discrepancies observed. For example, accumulating evidence may explain the discrepancy in recommendations for the predictive use of ERCC1 testing in NSCLC. In 2009, ASCO cited insufficient prospective phase III data for the predictive use of ERCC1 in NSCLC. Two years later, the NCCN gave it a similar recommendation and later upgraded the test to a category 2 recommendation in 2013. Another possible explanation is the different appraisal of evidence by different organizations. For CYP2D6 testing for tamoxifen use in breast cancer patients, the lone positive recommendation among four negative ones is based on expert consensus rather than systematic review of published evidence. This may also be the case for BRAF testing when considering anti-EGFR therapy in colon cancer, in which positive recommendations are issued at two different times by the NCCN, and a negative recommendation is issued by EGAPP at one of these times.
The circumstances of Oncotype DX suggest that the disagreements may be attributable to a combination of emerging evidence and different rating criteria. For prognostic and predictive purposes in lymph node–negative, ER-positive patients, there was only one negative recommendation among four other positive recommendations, suggesting different rating criteria. However, the recommendations span across all three recommendation categories over a 5-year period, with the most recent recommendation being category 1. This recommendation came from the National Institute for Health and Clinical Excellence, and, although category 1, it was noted that prospective confirmation was needed for the assay’s clinical utility.
For RRM1 and UGT1A1, the reasons are less obvious, partly because only two organizations commented on each. For RRM1 testing, there was an initial negative recommendation, followed by a moderate positive recommendation less than a year later. For UGT1A1, EGAPP found insufficient evidence to recommend for or against use. The NACB recommended testing as a useful adjunct for high-intensity irinotecan dosing, and it strongly recommends the use of testing according to the NACB rating scheme (level A), but the strength of the evidence on which the recommendation is based on can be “limited by the number, quality, or consistency of the individual studies; generalizability to routine practice; or indirect nature of the evidence.” On the other hand, in the EGAPP decision scheme to determine the clinical utility of a test, the decision to recommend a test is tied to the available evidence. Thus, EGAPP cited insufficient evidence because there were no prospective trials showing that UGT1A1 testing could avoid toxicity.
Others have noted the complexity and variability of grading evidence and health recommendations and have suggested a single system24 to standardize how genomic applications are recommended. Regardless of the reasons why there are different recommendations, the disagreements can be problematic when these tests are being considered for use in clinical practice. Clinicians treating patients may not always know which guidelines and recommendations to follow, especially when there are conflicting reports on the use of a genomic test.
There are some limitations of note in our empirical evaluation. First, we identified cancer genomic tests by beginning with recommendation statements issued by advisory groups, but in doing so may have introduced a bias in which only those tests that reached the point of discussion were included. This method may have left out previous tests that had already been implemented in practice long enough that no recommendation statements advocating their use was ever issued. Second, FDA approval may have influenced recommendations and vice versa. For example, one of the Blue Cross Blue Shield Technology Evaluation Center’s assessment criteria is approval from governmental regulatory bodies; therefore, not receiving FDA approval or clearance could have impacted a negative recommendation. Third, we did not consider the methods on which organizations based their guidelines, i.e., whether their methods were based on systematic review of the literature or a combination of literature review and expert consensus. Nevertheless, it was interesting to probe how different organizations with different approaches reached the same or different conclusions. Fourth, to make comparisons between recommendations, we aggregated recommendations so they fit into our three levels of recommendation. In doing so, the comparisons may not be completely fair and may have lost the nuances of the original recommendation. Fifth, in collecting historical recommendations, we relied on information listed on websites; it is possible that some organizations chose not to indicate whether previous recommendations were made. Sixth, we limited our definition of genetic testing so that they excluded protein markers, including those that are indicative of gene mutation status, such as HER2. Although this was performed to limit the scope of analysis and make comparisons fair, analysis of these markers may further our understanding. Finally, although efforts were made to encompass all expert recommendations in the field, we may have missed important potential sources, particularly guidelines issued by other countries. We should also point out that for laboratory-developed tests that fall under the jurisdiction of the Centers for Medicare and Medicaid Services, we thought Clinical Laboratory Improvement Amendments (CLIA) certification may be another facet influencing the trajectory of tests. Because there is no official, publicly available, dedicated CLIA database, we searched for information on the Genetic Testing Registry25 and known manufacturer and laboratory websites. However, there were concerns about the voluntary nature of information provided by the Genetic Testing Registry and the possibility of missed CLIA-certified testing providers. For these reasons, we did not incorporate CLIA certification as part of our analysis.
We should also acknowledge that the landscape of omics testing is rapidly changing and large-scale multiplexed genomic testing has been advocated, including routine whole-genome sequencing. However, the challenges with such approaches cannot be underestimated,26 and the exact role, if any, of such testing technologies is still unknown.
Allowing for these limitations, our review systematically summarizes the current translational landscape of genetic testing in oncology. Our findings indicate that only a few tests have strong recommendations and it is difficult to predict what kinds of tests will receive a favorable recommendation. For tests that have multiple organizations commenting on them, one-third had conflicting recommendations, and this may be a source of confusion for clinicians trying to decide the clinical appropriateness of these tests. We have found that the process of reaching some recommendation can take a long time from the discovery of the test. Many others have already proposed interesting ideas to accelerate translation of substantiated tests. The National Cancer Institute’s newly launched National Clinical Trials Network includes a series of large-scale clinical studies to find targeted therapies using next-general sequencing and high-throughput technologies.27 Ginsburg et al.28 proposed a rapid learning health-care model linked to comparative effectiveness research and personalized medicine by matching detailed patient data with health-outcomes data. Recent successes in tumor genome profiling and understanding tumor heterogeneity29 are promising. The extent to which such individualized, N-of-1 approaches may accelerate translation without increasing false-positive claims is an open question. Other than complications in the scientific processes, regulatory hurdles may add another element of translational stalling. This is especially true when more than one company is involved, as is the case for many companion diagnostic projects when there are diagnostic developers, pharmaceutical companies, and testing laboratories.30 The FDA has recently issued a special report on personalized medicine with specific plans to accelerate the development of promising new therapeutics and changes in regulatory processes that accommodate the specific challenges of personalized medicine products.31 Concerning the oversight of laboratory-developed tests, the document presents ongoing development of a risk-based framework to ensure safety and effectiveness of tests while encouraging innovation and progress.30 A draft proposal regarding the framework has been provided to Congress and as of August 2014 it has been available for public viewing on the FDA website.32 It remains to be seen whether and how these developments may accelerate reliable translation for cancer genomic tests.
Disclosure
The authors declare no conflict of interest.
References
Tremblay J, Hamet P . Role of genomics on the path to personalized medicine. Metabolism 2013;62(suppl 1):S2–S5.
Pasic MD, Samaan S, Yousef GM . Genomic medicine: new frontiers and new challenges. Clin Chem 2013;59:158–167.
Weiss RL . The long and winding regulatory road for laboratory-developed tests. Am J Clin Pathol 2012;138:20–26.
Teutsch SM, Bradley LA, Palomaki GE, et al.; EGAPP Working Group. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP)Initiative: methods of the EGAPP Working Group. Genet Med 2009;11:3–14.
National Institute for Health and Clinical Excellence. The Guidelines Manual. 2013. http://www.nice.org.uk/guidelinesmanual. Accessed 31 October 2013.
American Society of Clinical Oncology. ASCO Guidelines: Methodology Manual. 2013. http://www.asco.org/sites/default/files/methodology_manual_1.25.11_0.pdf. Accessed 5 November 2013.
BlueCross BlueShield Association. Technology Evaluation Center (TEC). 2013. http://www.bcbs.com/blueresources/tec/.
US Preventive Services Task Force. Methods and Processes. 2013. http://www.uspreventiveservicestaskforce.org/methods.htm. Accessed 5 November 2013.
National Comprehensive Cancer Network. NCCN Guidelines and Derivative Information Products: User Guide. 2013. http://www.nccn.org/professionals/default.aspx. Accessed 5 November 2013.
Agency for Healthcare Research and Quality. National Guideline Clearinghouse. 2013. http://www.guideline.gov/index.aspx. Accessed 31 October 2013.
Febbo PG, Ladanyi M, Aldape KD, et al. NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. J Natl Compr Canc Netw. 2011;9(suppl 5):S1–32; quiz S33.
US Department of Health and Human Services. US Food and Drug Administration. 2013. www.fda.gov. Accessed 5 November 2013.
Contopoulos-Ioannidis DG, Alexiou GA, Gouvias TC, Ioannidis JP . Medicine. Life cycle of translational research for medical interventions. Science 2008;321:1298–1299.
Contopoulos-Ioannidis DG, Ntzani E, Ioannidis JP . Translation of highly promising basic science research into clinical applications. Am J Med 2003;114:477–484.
National Cancer Institute. Common Cancer Types. 2013. http://www.cancer.gov/cancertopics/types/commoncancers. Accessed 5 November 2013.
Altman DG, Riley RD . Primer: an evidence-based approach to prognostic markers. Nat Clin Pract Oncol 2005;2:466–472.
Riley RD, Sauerbrei W, Altman DG . Prognostic markers in cancer: the evolution of evidence from single studies to meta-analysis, and beyond. Br J Cancer 2009;100:1219–1229.
Morris ZS, Wooding S, Grant J . The answer is 17 years, what is the question: understanding time lags in translational research. J R Soc Med 2011;104:510–520.
Poste G . Bring on the biomarkers. Nature 2011;469:156–157.
Bollag G, Tsai J, Zhang J, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 2012;11:873–886.
Elion GB, Hitchings GH, Vanderwerff H . Antagonists of nucleic acid derivatives. VI. Purines. J Biol Chem 1951;192:505–518.
Weinshilboum RM, Sladek SL . Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet 1980;32:651–662.
van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530–536.
Guyatt GH, Oxman AD, Vist GE, et al.; GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924–926.
National Center for Biotechnology Information. Genetic Testing Registry (GTR). 2013. http://www.ncbi.nlm.nih.gov/gtr/. Accessed 5 November 2013.
Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA 2014;311:1035–1045.
Abrams J, Conley B, Mooney M, et al. National Cancer Institute’s precision medicine initiatives for the new National Clinical Trials Network. Am Soc Clin Oncol Educ Book. 2014; 71–76.
Ginsburg GS, Kuderer NM . Comparative effectiveness research, genomics-enabled personalized medicine, and rapid learning health care: a common bond. J Clin Oncol 2012;30:4233–4242.
Patel L, Parker B, Yang D, Zhang W . Translational genomics in cancer research: converting profiles into personalized cancer medicine. Cancer Biol Med 2013;10:214–220.
Drucker E, Krapfenbauer K . Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J 2013;4:7.
Food and Drug Administration. Personalized Medicine: FDA’s Unique Role and Responsibilities in Personalized Medicine. 2014. http://www.fda.gov/scienceresearch/specialtopics/personalizedmedicine/default.htm. Accessed 10 February 2014.
Food and Drug Administration. Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs). 2014. http://www.fda.gov/downloads/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/UCM407409.pdf. Accessed 7 August 2014.
Acknowledgements
We acknowledge W. David Dotson and Andrew N. Freedman for their thoughtful comments on the manuscript. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention, the National Cancer Institute, or the US Department of Health and Human Services.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 112 kb)
Supplementary Table S2
(DOC 42 kb)
Supplementary Table S3
(DOC 557 kb)
Rights and permissions
About this article

Cite this article
Chang, C., Tingle, S., Filipski, K. et al. An overview of recommendations and translational milestones for genomic tests in cancer. Genet Med 17, 431–440 (2015). https://doi.org/10.1038/gim.2014.133
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.133
