
Abstract
Prader-Willi syndrome is characterized by severe infantile hypotonia with poor suck and failure to thrive; hypogonadism causing genital hypoplasia and pubertal insufficiency; characteristic facial features; early-childhood onset obesity and hyperphagia; developmental delay/mild intellectual disability; short stature; and a distinctive behavioral phenotype. Sleep abnormalities and scoliosis are common. Growth hormone insufficiency is frequent, and replacement therapy provides improvement in growth, body composition, and physical attributes. Management is otherwise largely supportive. Consensus clinical diagnostic criteria exist, but diagnosis should be confirmed through genetic testing. Prader-Willi syndrome is due to absence of paternally expressed imprinted genes at 15q11.2-q13 through paternal deletion of this region (65–75% of individuals), maternal uniparental disomy 15 (20–30%), or an imprinting defect (1–3%). Parent-specific DNA methylation analysis will detect >99% of individuals. However, additional genetic studies are necessary to identify the molecular class. There are multiple imprinted genes in this region, the loss of which contribute to the complete phenotype of Prader-Willi syndrome. However, absence of a small nucleolar organizing RNA gene, SNORD116, seems to reproduce many of the clinical features. Sibling recurrence risk is typically <1%, but higher risks may pertain in certain cases. Prenatal diagnosis is available.
Genet Med 2012:14(1):10–26.
Similar content being viewed by others


Overview
Prader-Willi syndrome (PWS) is a multisystem disorder with an estimated prevalence in several studied populations of 1/10,000–1/30,000. It is characterized by severe hypotonia with poor suck and feeding difficulties in early infancy, followed in later infancy or early childhood by excessive eating and gradual development of morbid obesity unless eating is externally controlled. Motor milestones and language development are delayed, and all individuals have some degree of cognitive disability. A distinctive behavioral phenotype is common, with temper tantrums, stubbornness, and manipulative and compulsive behaviors. Hypogonadism is present in both males and females and manifests as genital hypoplasia, incomplete pubertal development, and, in most, infertility. Short stature is common, related to growth hormone (GH) insufficiency. Characteristic facial features, strabismus, and scoliosis are often present, and there is an increased incidence of sleep disturbance and type II diabetes mellitus, the latter particularly in those who become obese. The physical features and impact of treatment are illustrated in Figures 1 and 2 .

(a) An 8-month-old female with hypotonia, hypogonadism, and need for assisted feeding. (b) A 19-year-old male with inadequate dietary control (body mass index (BMI) = 67; Z score = +3.49) showing typical body habitus of Prader-Willi syndrome (PWS) with fat distributed primarily in abdomen, hips, and thighs. (c) A 34-year-old man in relatively good dietary control (BMI = 30; Z score = +1.66) living in a specialized PWS group home. Note the hanging skin left from his history of morbid obesity. Informed consent was obtained for publication of these photographs.

(a and b) Seven- and 13-year-old children, respectively, not receiving growth hormone treatment. (c and d) Seven- and 13-year-old children, respectively, who have had growth hormone treatment and good weight control. Informed consent was obtained for publication of these photographs.
PWS occurs as the result of absence of expression of paternal genes from chromosome 15q11.2-q13. A number of genes in this region are subject to genomic imprinting and are normally active only from the paternally contributed chromosome 15; those same alleles from the maternally contributed chromosome 15 are inactivated by epigenetic factors and are not expressed. The absence of expression of one or more of the paternally inherited genes must contribute to the phenotype of PWS. This lack of expression occurs by three primary mechanisms: (i) deletion of a 5–6 Mb region from the paternally contributed chromosome 15 (found in 65–75% of affected individuals); (ii) maternal uniparental disomy (UPD) 15 (found in 20–30%); and (iii) a defect in the genomic region that controls the imprinting process, a so-called imprinting defect (ID; 1–3%). IDs are usually sporadic but can be due to a microdeletion in the imprinting center (IC) and in the latter case may be inherited. Although published consensus clinical diagnostic criteria are available and accurate, the mainstay of diagnosis is genetic testing. DNA-based methylation testing will detect abnormal parent-specific imprinting within the Prader-Willi critical region on chromosome 15; this testing determines whether the region is maternally inherited only (i.e., the normal paternal imprint is absent) and detects more than 99% of affected individuals. Diagnosis can also be made by fluorescence in situ hybridization (FISH) or chromosomal microarray (CMA) for deletion 15q11.2-q13, DNA polymorphism analysis in parents and the affected individual for UPD, and an experienced referral laboratory for testing for an IC microdeletion. This genetic testing is important to confirm the diagnosis of PWS in all individuals, but especially so in those who have atypical findings or are too young to manifest sufficient features to make the diagnosis with certainty on clinical grounds.
Manifestations and Natural History
Prenatal characteristics
The birth weight, length, and body mass index (BMI) of infants with PWS are 15–20% smaller than those of their unaffected siblings (although often still in the normal range), indicating that growth is abnormal prenatally.1,2,3 Prenatal hypotonia usually results in decreased fetal movement, abnormal fetal position at delivery, and increased incidence of assisted delivery or cesarean section.1
Hypotonia
Infantile hypotonia ( Figure 1a ) is a nearly universal finding, causing decreased movement and lethargy with decreased spontaneous arousal, weak cry, and poor reflexes, including a poor suck. The hypotonia is central in origin, and neuromuscular studies including muscle biopsy, when done for diagnostic purposes, are generally normal or show nonspecific signs of disuse. The poor suck and lethargy result in failure to thrive in early infancy, and gavage feeding or the use of special nipples is generally required for a variable period of time, usually weeks to months.4 By the time that the child is drinking from a cup or eating solids, a period of approximately normal eating behavior occurs. The hypotonia improves over time, but adults remain mildly hypotonic with decreased muscle bulk and tone.
Developmental disability
Delayed motor development is present in 90–100% of children with PWS, with average early milestones achieved at about double the normal age (e.g., sitting at 12 months and walking at 24 months). Language milestones are also typically delayed. Intellectual disabilities are generally evident by the time the child reaches school age. Testing indicates that most individuals with PWS fall in the mild intellectual disability range (mean intelligence quotient [IQ]: 60–70), with approximately 40% having borderline or low-normal intelligence and approximately 20% having moderate intellectual disability. Regardless of measured IQ, most children with PWS have multiple severe learning disabilities and poor academic performance for their mental abilities.5 Although a small proportion of affected individuals have extremely impaired language development, verbal ability is a strength for most; however, articulation abnormalities are frequent.
Hyperphagia and obesity
Nutritional phases. In contrast to the long-held view that there are two distinct nutritional phases in PWS, failure to thrive followed by “hyperphagia leading to obesity,” a recent collaborative study found that the transition between nutritional phases is much more complex, with seven different nutritional phases through which individuals with PWS typically progress3 ( Table 1 ). Phase 0 occurs in utero, with decreased fetal movements and growth restriction compared with unaffected siblings. In Phase 1, the infant is hypotonic and not obese, with subphase 1a characterized by difficulty feeding with or without failure to thrive (ages: birth to 15 months; median age at completion: 9 months). This phase is followed by subphase 1b when the infant grows steadily along a growth curve, and weight is increasing at a normal rate (median age of onset: 9 months; range: 5–15 months). Phase 2 is associated with weight gain; in subphase 2a, the weight increases without a significant change in appetite or caloric intake (median age of onset: 2.08 years), whereas in subphase 2b, the weight gain is associated with a concomitant increased interest in food (median age of onset: 4.5 years). Phase 3 is characterized by hyperphagia, typically accompanied by food seeking and lack of sense of satiety (median age of onset: 8 years). Not all individuals with PWS go through all these stages, but the majority do. In addition, some adults progress to Phase 4, which is when an individual who was previously in Phase 3 no longer has an insatiable appetite and is able to feel full.
Obesity. The hyperphagia that occurs in Phase 3 is believed to be caused by a hypothalamic abnormality resulting in lack of satiety. Food-seeking behavior, with hoarding or foraging for food, eating of inedibles, and stealing of food or money to buy food are common. In most, gastric emptying is delayed, and vomiting is rare. Obesity results from these behaviors and from decreased total caloric requirement. The latter is due to decreased resting energy expenditure resulting from decreased activity and decreased lean body mass (primarily muscle) compared with unaffected individuals.6 The obesity in PWS is primarily central (abdomen, buttocks, and thighs) in both sexes, and interestingly, there is less visceral fat in obese individuals than would be expected for the degree of obesity.7 Obesity and its complications are the major causes of morbidity and mortality (see “Morbidity and Mortality”).
Several independent groups have shown that ghrelin levels are significantly elevated in hyperphagic older children and adults with PWS before and after meals.8,9,10,11,12 Ghrelin is a potent circulating orexigenic hormone that is produced mainly in the stomach. Circulating ghrelin levels rise after fasting and are suppressed by food intake. The appetite-inducing effect acts through the appetite regulating pathway in the hypothalamus. Ghrelin levels are lower in non-PWS obese individuals versus lean controls and they decrease with age.13
In a small study of nine nonhyperphagic children with PWS (17–60 months of age), similar levels of circulating ghrelin as in the eight control children matched for BMI, age, and sex were found.14 By contrast, in a larger and younger study cohort of 40 children and adolescents with PWS (range: 0.2–17.2 years, median age: 3.6 years), ghrelin levels were significantly elevated in the PWS group at any age compared with 84 age and BMI-matched controls.15 In fact, the highest ghrelin levels in PWS were found in the youngest children. Thus, in their study, the hyperghrelinemia was occurring long before the development of obesity and increased appetite in PWS. Furthermore, several groups have now shown that pharmacological reduction of ghrelin to normal levels in PWS, using either short or long acting agents, did not affect the weight, appetite, or eating behavior in hyperphagic individuals.16,17,18 At this time, there are no consistently identified hormonal abnormalities to explain the hyperphagia, and the metabolic correlates of hyperphagia in PWS are still uncertain.
Dysmorphic features
Characteristic facial features include narrow bifrontal diameter, almond-shaped palpebral fissures, narrow nasal bridge, and thin upper vermillion with down-turned corners of the mouth ( Figures 1 and 2 ). These may or may not be apparent at birth and slowly evolve over time.
The hands are slender with a hypoplastic ulnar bulge, and in young children, the dorsa of the palm and fingers may be puffy and the fingers may appear tapered. Hypopigmentation of hair, eyes, and skin are common in subjects with a deletion due to a concomitant loss of one copy of the OCA2 gene. (A homozygous loss of the OCA2 gene results in tyrosinase-positive albinoidism.)
Behavioral and psychiatric disturbance
A characteristic behavior profile with temper tantrums, stubbornness, controlling and manipulative behavior, compulsivity, and difficulty with change in routine becomes evident in early childhood in 70–90% of individuals with PWS.19 Some of the behavioral characteristics are suggestive of autism; in one study, 19% of 59 individuals with PWS vs. 5% of age-, sex-, and IQ-matched controls satisfied diagnostic criteria for autism.20 In another study of 58 children, attention deficit/hyperactivity symptoms and insistence on sameness were common and of early onset.21 This behavior disorder has been reported to increase with age and BMI,22 although it diminishes considerably in older adults.23 Psychosis is evident by young adulthood in 10–20% of affected individuals.24,25,26 Behavioral and psychiatric problems interfere most with the quality of life in adolescence and adulthood.
Hypogonadism
In both sexes, hypogonadism is present and manifests as genital hypoplasia, incomplete pubertal development, and infertility in the majority. Genital hypoplasia is evident at birth and throughout life. In males, the penis may be small, and most characteristic is a hypoplastic scrotum that is small, poorly rugated, and poorly pigmented. Unilateral or bilateral cryptorchidism is present in 80–90% of males. In females, the genital hypoplasia is often overlooked; however, the clitoris and labia, especially the labia minora, are generally small from birth. The hypogonadism causes incomplete, delayed, and sometimes disordered pubertal development. Precocious adrenarche occurs in approximately 15–20% in both sexes. Primary amenorrhea or oligomenorrhea are present in females. Infertility is the rule in both sexes, although a few instances of reproduction in females have been reported27,28 and presented (Vats and Cassidy, unpublished data). The largest recent study of hypogonadism, which included 84 individuals with PWS (half males and half females) ages 2–35 years,29 identified the following frequencies: in males: cryptorchidism 100%, small testes 76%, and scrotal hypoplasia 69%; in females: labia minora and/or clitoral hypoplasia 76%, primary amenorrhea 56%, spontaneous menarche (mostly spotting) 44% of those older than 15 years; in both sexes: premature pubarche 14%, and precocious puberty 3.6% (one male and two females). Although the hypogonadism in PWS has long been believed to be entirely hypothalamic, resulting in low gonadotropins and subsequent low gonadal hormones, recent studies have suggested that there is a combination of hypothalamic and primary gonadal deficiencies.30,31,32 That conclusion was largely based on absence of hypogonadotropism and abnormally low inhibin B in both sexes.
Short stature and growth hormone deficiency
Short stature may be apparent in childhood and is almost always present by the second decade in the absence of GH replacement, and lack of a pubertal growth spurt results in an average untreated height of 155 cm for males and 148 cm for females. The hands and feet grow slowly and are generally below the fifth percentile by age 10 years. Data from at least 15 studies involving more than 300 affected children document reduced GH secretion in PWS.33 Best practice in early intervention for PWS includes recommendations for GH therapy.4,34 GH therapy decreases fat mass and increases muscle mass ( Figure 2 ). Preliminary data also suggest that it may have a beneficial effect on weight gain, and possibly appetite, in individuals with PWS.33,35 Infants with PWS treated with GH therapy have improvements in head circumference, height, BMI, body composition (with improvement of lean muscle mass and delay of fat tissue accumulation), body proportions, acquisition of gross motor skills, language acquisition, and cognitive scores.36,37,38,39,40,41,42,43 Older children and adolescents treated with GH therapy not only have the aforementioned physical benefits of the treatment but are also reported to have improvements in behaviors with lack of behavioral deterioration during adolescence.44 Recent studies have found that up to 72% of children with PWS treated with GH therapy have a serum insulin-like growth factor 1 (IGF-1) level that is >2 standard deviations after 24 months of treatment despite lower doses of GH than are used for children with isolated GH deficiency, which raises concerns about safety issues related to high IGF-1 levels in this population.39 However, IGF-1 to IGF binding protein 3 ratios remained stable with GH therapy, suggesting that bioavailable IGF-1 is not elevated to a greater extent than is seen in individuals with other causes of GH deficiency.
GH deficiency is also seen in adults with PWS, although study findings differ on the prevalence in the adult population.45,46 One study found that adults with PWS due to maternal UPD had lower GH secretion than those with deletion, although these results have not been replicated.47 Several studies have documented the safety and efficacy of GH treatment in adults with PWS on body composition and quality of life.48,49 One study found that the beneficial effects of GH therapy were unrelated to the pretreatment GH or serum IGF-1 levels.49
Concern about the possible contribution of GH administration to unexpected death has been raised by reported deaths of individuals within a few months of starting GH therapy.50,51,52,53,54,55 The few reported deaths were mostly in obese individuals who had preexisting respiratory or cardiac disorders with evidence of upper airway obstruction and uncorrected tonsillar and adenoidal hypertrophy. In the database of one pharmaceutical company, five of 675 children treated with GH died suddenly of respiratory problems.56 In another study, the rate of death in affected individuals on and off GH did not differ.57 A study of the natural history of PWS in one region of the United Kingdom found the overall death rate of individuals with PWS to be as high as 3% per year without GH therapy.58 Thus, the relationship of GH administration to unexpected death remains unclear. However, a recent long-term study of 48 treated children suggests that the benefits of treatment exceed the risks.42
Other endocrine issues
Central adrenal insufficiency. Central adrenal insufficiency (CAI) following overnight single-dose metyrapone tests was noted in 60% of children with PWS in one study, suggesting that this may be the cause of the high incidence of sudden death in this population.59 It is known that introducing GH therapy can precipitate adrenal crisis in individuals with incipient adrenal insufficiency by accelerating the peripheral metabolism of cortisol, which may explain the correlation between the incidence of sudden death at the beginning of GH treatment and CAI in individuals with PWS.60 However, two subsequent studies have found normal cortisol responses to low- and high-dose synacthen testing, as well as to insulin tolerance testing,61,62 so whether CAI is a true issue for individuals with PWS remains uncertain at this time and there is no consensus among endocrinologists as to whether evaluation for CAI should be performed on every patient with PWS or just those with symptoms consistent with adrenal insufficiency.
Hypothyroidism. Central hypothyroidism, with a normal thyroid stimulating hormone value and low free thyroxine level, has been documented in up to 25% of individuals with PWS, with a mean age of diagnosis and treatment of 2 years.63,64
Impaired glucose tolerance and diabetes mellitus. Up to 25% of adults with PWS (particularly those with significant obesity) have noninsulin-dependent diabetes mellitus65 with a mean age of onset of 20 years. A study of a large French cohort with PWS (ages: 2–18.8 years) revealed the presence of impaired glucose tolerance in 4% of individuals (mean age: 10.2 years) but no diabetes mellitus in those younger than 20 years of age.64
Other findings
-
Sleep abnormalities are well documented and include reduced rapid eye movement (REM) latency, altered sleep architecture, oxygen desaturation, and both central and obstructive apnea.66,67 Primary hypothalamic dysfunction is thought to be the cause of the alterations in sleep microstructure and abnormalities in ventilation during sleep, with studies showing low levels of orexin and hypocretin in the cerebrospinal fluid and decreased levels of acetyl-cholinergic neurons in the pedunculo-pontine tegmental nucleus.68,69,70,71 Some individuals with PWS have excessive daytime sleepiness, which resembles narcolepsy, with rapid onset of REM sleep and decrease in non-REM sleep instability.70
-
Strabismus is seen in 60–70%.
-
Scoliosis, present in 40–80%, varies in age of onset and severity. Randomized trials have found no relationship between GH therapy and the age of onset or severity of scoliosis in children with PWS.40
-
Up to 50% of affected individuals may have recurrent respiratory infections. There are no reports of immune deficiency in PWS, and the increase in respiratory infection may be related to respiratory muscle hypotonia and thus decreased cough.
-
Rates of the following are increased: Bone fractures caused by osteopenia Leg edema and ulceration (especially in the obese) Skin picking Altered temperature sensation Decreased saliva flow High vomiting threshold Seizures (10–20%)
Neuroimaging
In a recent study, all 20 individuals with PWS who were evaluated had brain abnormalities that were not found in 21 sibs or 16 individuals with early-onset morbid obesity who did not have PWS.74 All had ventriculomegaly; 50% had decreased volume of brain tissue in the parietal-occipital lobe; 60% had Sylvan fissure polymicrogyria; and 65% had incomplete insular closure. In another study, these authors reported white matter lesions in some people with PWS.75 A study of brain MRIs from 91 individuals with PWS from another group showed reduced pituitary height in 49% and some neuroradiologic abnormality in 67%.76 The implications of these findings are unknown.
Morbidity and Mortality
The mortality rate in PWS is higher than in controls with intellectual disability, with obesity and its complications being factors.77 Based on a population study, the death rate has been estimated at 3% per year.65 Another large study suggested a sixfold relative risk of death in PWS versus other developmentally disabled individuals.77 Two multicenter series of individuals who died of PWS have been reported,78,79 and an extensive case and literature review of 64 cases of death in PWS was done.80 Respiratory and other febrile illnesses were the most frequent causes of death in children, and obesity-related cardiovascular problems and gastric causes or sleep apnea were most frequent in adults. Other causes of morbidity include diabetes mellitus, thrombophlebitis, and skin problems (e.g., chronic edema and infection from skin picking).
A few individuals have been reported to have respiratory or gastrointestinal infections resulting in unexpected death; of these, three who died as a result were noted to have small adrenal glands,79 although this is not a common finding. The possibly increased incidence of CAI in PWS may provide an explanation for some of these unexpected and sudden deaths.
Acute gastric distention and necrosis have been reported in a number of individuals with PWS,81,82 particularly following an eating binge among those who are thin but were previously obese. It may be unrecognized because of high pain threshold and can be fatal.
Choking, especially on hot dogs, has been reported as a cause of death in approximately 8% of deaths in individuals with PWS.83
Clinical Diagnosis
Consensus diagnostic criteria
Consensus clinical diagnostic criteria for PWS using a numerical scale ( Table 2 ), developed in 199384 before the availability of diagnostic testing, have proven to be accurate.85 However, confirmation of diagnosis requires molecular genetic testing.
The following additional descriptions pertain to the diagnostic criteria. The hypotonia is central and improves with age. The feeding problems are related to poor suck and usually result in the need for gavage feeding or other special feeding techniques. Cryptorchidism and hypoplastic scrotum are very common in males, pubertal development is incomplete, and infertility is almost universal. Developmental disability is usually mild to moderate. The decreased movement and lethargy in infancy improve with age. Typical behavior problems include temper tantrums, obsessive-compulsive behavior, stubbornness, rigidity, stealing, and lying. A number of supportive findings were also reported in the consensus criteria for clinical diagnosis, although they do not add points to the diagnosis score. These include high pain threshold, decreased vomiting, scoliosis and/or kyphosis, early adrenarche, osteoporosis, unusual skill with jigsaw puzzles, and normal neuromuscular studies (muscle biopsy, electromyography and nerve conduction velocity), if tested.
Findings prompting genetic testing
Subsequent to the availability of genetic testing, findings that should prompt diagnostic testing were proposed based on analysis of satisfied diagnostic criteria in individuals in whom the diagnosis of PWS has been molecularly confirmed.85 These differ by age group and indicate that the presence of the following findings is sufficient to justify DNA methylation analysis for PWS (see “Diagnostic Testing”):
Birth to age 2 years: Hypotonia with poor suck in the neonatal period.
Age 2–6 years: Hypotonia with history of poor suck and global developmental delay.
Age 6–12 years: History of hypotonia with poor suck (hypotonia often persists), global developmental delay, and excessive eating with central obesity if diet is uncontrolled.
Age 13 years to adulthood: Cognitive impairment (usually mild intellectual disability), excessive eating with central obesity (if caloric intake is uncontrolled), hypothalamic hypogonadism, and characteristic behavior problems.
Genetics of PWS
Molecular genetics
The PWS region is localized to a 5–6 Mb genomic region on the proximal long arm of chromosome 15 (15q11.2-q13). It lies within a smaller 2.5 Mb differentially imprinted region. PWS is a contiguous gene disorder, as studies thus far indicate that the complete phenotype is due to the loss of expression of several genes. It is also an example of an imprinted condition, as the expression of relevant genes in the 15q11.2-q13 region is dependent on parental origin.86,87
PWS in conjunction with Angelman syndrome (AS) represent perhaps the best examples of genomic imprinting in humans. The majority of the genes in mammals display a Mendelian pattern of expression where normal alleles inherited from each parent are equally expressed. By contrast, probably <1% of our genes are imprinted, having an expression pattern determined by the parent of origin. Although the DNA sequence of the imprinted maternally and paternally inherited alleles is the same, multiple epigenetic factors (such as DNA methylation, histone modifications, and chromatin conformation) ultimately will determine whether the imprinted allele is expressed or repressed.88,89
The genomic and epigenetic changes causing PWS all lead to a loss of expression of the normally paternally expressed genes on chromosome 15q11.2-q13. Absence of the paternally inherited copy of these genes, or failure to express them, causes total absence of expression for those genes in the affected individual because the maternal contribution for these genes has been programmed by epigenetic factors to be silenced.86 Conversely, a loss of expression of the preferentially maternally expressed UBE3A gene in this region by several different possible mechanisms leads to AS.90,91
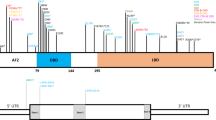
Genes in the 15q11.2-q13 region. The 15q11.2-q13 region can be roughly divided into four distinct regions that are delineated by three common deletion breakpoints,92 which lie within segmental duplications93 ( Figure 3 ): (1) a proximal nonimprinted region between the two common proximal breakpoints (BP1 and BP2) containing four biparentally expressed genes, NIPA1, NIPA2, CYF1P1, and GCP5.94 (2) The “PWS paternal-only expressed region” containing five polypeptide coding genes (MKRN3, MAGEL2, NECDIN, and the bicistronic SNURF-SNRPN); C15orf2 (an intronless gene that is biallelically expressed in testis but only expressed from the paternal allele in brain); a cluster of C/D box small nucleolar RNA genes (snoRNAs); and several antisense transcripts (including the antisense transcript to UBE3A). (3) The “Angelman syndrome (AS) region” containing the preferentially maternally expressed genes UBE3A and ATP10A. (4) A distal nonimprinted region containing a cluster of three GABA receptor genes, the gene for oculocutaneous albinism type 2 (OCA2), HERC2, and the common distal breakpoint (BP3).

Summary of the genetic and expression map of chromosomal region 15q11.2-q13. The Prader-Willi syndrome (PWS) region (shown in blue) has five paternal-only (PWS region) expressed unique copy genes that encode polypeptides (MKRN3, MAGEL2, NECDIN, and SNURF-SNRPN) and a family of six paternal-only expressed snoRNA genes. Only UBE3A and ATP10A (shown in orange), related to Angelman syndrome (AS), have preferential maternal-only expression, and this imprinted expression is limited to certain tissue specific regions (specifically brain). The bipartite imprinting center (IC) lies proximal to the bicistronic SNURF-SNRPN gene and within the 2.5 Mb PWS/AS imprinted region. The cluster of GABA receptor genes (GABRB3, GABRA5, and GABRG3), OCA2 (type II albinism), and HERC2 are not imprinted and have biparental expression (shown in green). The jagged vertical lines denote the three common 5–6 Mb PWS and AS deletion breakpoints, BP1, BP2, and BP3. On rare occasions, there will be a distal breakpoint at BP4 or BP5. In between BP1 and BP2 lie four additional, nonimprinted genes, NIPA1, NIPA2, CYFIP1, and GCP5.94 Type 1 deletions (T1D) extend from BP1 to BP3, and type 2 deletions (T2D) extend from BP2 to BP3. Note that there are more copies of the SNORD116 and SNORD115 genes than are shown, and the map has not been precisely drawn to scale.
Central to the PWS region is the SNURF-SNRPN gene. It is a bicistronic gene encoding two different proteins. Exons 4–10 were described first and encode the protein SmN, which is a spliceosomal protein involved in mRNA splicing.95 SNURF is encoded by exons 1–3, which produces a polypeptide of unknown function.96 At the 5′ end of the SNURF-SNRPN gene is a CpG island encompassing the promoter, exon 1, and intron 1. This is a differentially methylated region, which is unmethylated on the paternally inherited expressed allele and methylated on the maternally inherited repressed allele.95 The promoter and exon 1 overlap with the paternal PWS IC.97 The SNURF-SNRPN gene also serves as the host for the six snoRNA genes located telomerically, which are regulated by the expression of SNURF-SNRPN. The UBE3A antisense transcript also arises from transcription of SNURF-SNRPN and is thought to lead to the repression of the paternally inherited UBE3A gene in humans and mice.98,99,100
The snoRNAs are present in single copy except for SNORD116 (previously named HBII-85) and SNORD115 (previously named HBII-52), which are present in 29 and 42 copies, respectively. It is thought that the snoRNAs are probably involved in the modification of mRNA by alternative splicing and that each snoRNA gene might have multiple targets. However, at the present time, only one target for a snoRNA gene (i.e., SNORD115) has been found and that is the serotonin 2C receptor.101 No targets have yet been found for SNORD116.
The exact function of each of the genes in determining the PWS phenotype remains to be elucidated, although possible insight has been gained by work with mouse models by multiple investigators. No single gene mutation has been found in humans that will explain all the features of PWS, unlike the situation in AS where single gene mutations of UBE3A fulfill all the major clinical criteria for AS.90,91 However, a “key” region to explain much of the PWS phenotype has been narrowed to the SNORD116 snoRNA gene cluster by several unique deletion and translocation families (reviewed by Buiting ref. 102). A crucial role for the SNORD115 locus was eliminated by an AS family with a familial microdeletion that included the entire SNORD115 gene cluster and the UBE3A locus.103 There was no obvious phenotype when this microdeletion was passed paternally, but it resulted in AS when inherited maternally.
Recently, there have been three separate reports of three different individuals with overlapping microdeletions (175–236 kb) that all encompass the SNORD116 gene cluster.104,105,106 All three have multiple clinical features typical of PWS including neonatal hypotonia, infantile feeding problems, rapid weight gain by 2 years of age, hyperphagia, hypogonadism, developmental delay/intellectual disability, and speech and behavioral problems. However, these three individuals also have features not typical of classical PWS, including tall stature as a child, large head circumference, lack of a “PWS facial gestalt,” and hand features not typical of PWS. Furthermore, rigorous neurobehavioral studies have not been performed to determine whether these individuals have the typical PWS behavioral phenotype. Nonetheless, it is clear from these studies that absence of the paternally derived SNORD116 gene cluster plays a major role in the PWS phenotype.
Molecular classes of PWS. There are three main molecular mechanisms that result in PWS: paternal deletion, maternal UPD 15, and ID ( Figure 4 ).

Genetic classes of Prader-Willi syndrome (PWS) and their average frequencies (I: deletion, II: uniparental disomy, III: imprinting defect). Bars represent chromosome 15. P indicates the paternally inherited chromosome 15, and M indicates the maternally contributed one. Class III occurs when there is biparental inheritance, but the paternally inherited chromosome 15 is imprinted in the manner typical of the maternal chromosome 15 (i.e., relevant genes are not expressed).
Paternal deletion. Most cases of PWS result from an interstitial microdeletion of the paternally inherited 15q11.2-q13 region.86,107,108 Deletions account for 65–75% of the individuals with PWS. The majority of individuals with deletions have one of two common proximal breakpoints (BP1 or BP2) and a common distal breakpoint (BP3).92,93 These recurrent common interstitial deletions measure approximately 5–6 Mb in size and are due to the presence of multiple copies of tandemly repeated sequences at the common breakpoints (BP1, BP2, and BP3) flanking the deleted region. These low copy repeat sequences stretch for approximately 250–400 kb and can cause nonhomologous pairing and aberrant recombination of the 15q11.2-q13 region during meiosis, leading to deletions (causing PWS or AS depending on parental origin), duplications (both maternal and paternal), triplications, and inverted dup (15).93,109,110,111,112 In addition, approximately 8% of those with a deletion have a unique or atypical sized deletion (i.e., not type 1 or 2) from a variety of etiologies, including an unbalanced translocation (Driscoll and Kim, unpublished data). A deletion that is smaller or larger than typically seen in PWS may affect the phenotype.
Maternal UPD. Maternal UPD 15 is the situation in which there are two chromosomes 15 from the mother and none from the father.113,114 This accounts for approximately 20–30% of individuals with PWS. Maternal UPD has been shown to be associated with advanced maternal age.3,115,116 A two-step process explains most cases of maternal UPD. First, there is a maternal nondisjunction event resulting in an ovum disomic for chromosome 15. After fertilization with a normal sperm, the resultant trisomy 15 in the blastocyst is lethal unless a second event produces a daughter cell that has lost one of the three chromosomes 15. This process, referred to as “trisomy rescue,” has been documented to result in PWS on a number of occasions based on the presence of a trisomic cell line in the placenta with UPD in the embryo.117,118,119,120 PWS results when the sole paternal 15 is lost, leaving two maternal 15s. Other mechanisms could also lead to UPD but seem to be much less common in PWS. A nullisomic cell line with only the maternal 15 could also result from nondisjunction, and a second event could result in duplication of this chromosome (“monosomy rescue”). In this case, all the genes would be identical in both alleles (isodisomy), and there is a risk of duplication of a deleterious gene in addition to PWS, which has been documented to occur for several genes on chromosome 15. There are other possible causes of UPD, including a postfertilization error (by somatic recombination or gene conversion), gametic complementation, and somatic replacement of a derivative chromosome. Trisomy associated with Robertsonian translocations may also resolve to disomy through loss of a chromosome and would result in UPD in 50% of cases. UPD can also be associated with small supernumerary chromosome 15 markers, and both maternal and paternal UPD 15 have been identified from this situation, although maternal is more common.121 The parental origin of these small markers is frequently unknown due to the small size and lack of unique genetic material. It has been estimated that approximately 5% of small supernumerary markers are associated with UPD.122
Imprinting defect. This molecular class affects the imprinting process on the paternally inherited chromosome 15. It only accounts for approximately 1–3% of individuals with PWS. Most IDs result from epigenetic causes (epimutations) and demonstrate a maternal-only DNA methylation pattern despite the presence of both parental alleles (i.e., biparental inheritance). DNA sequence changes are not found in these epimutations, and they are thought to be random errors in the imprinting process during spermatogenesis of the fathers86 or in early embryogenesis in the rare cases of somatic mosaicism.102 However, approximately 15% of individuals with an ID are found to have a very small deletion (7.5 to >100 kb) in the PWS IC region located at the 5′ end of the SNRPN gene and promoter (i.e., an IC deletion).97 Of these, about half have been inherited from an unaffected father with the IC deletion on his maternally inherited chromosome 15. The other half are de novo IC deletions on the paternally inherited 15 that arose during spermatogenesis in the father or after fertilization.88,123
Genotype-Phenotype Correlations
There are no features known to occur exclusively in individuals with one of the genetic classes. However, there are some statistical differences in the frequency or severity of some features between the two largest classes (deletion 15q11.2-q13 and UPD). Post-term delivery is more common with UPD.1 Individuals with UPD are less likely to have the hypopigmentation,115,124 typical characteristic facial appearance,115,116 or skill with jigsaw puzzles.125 In most studies, those with UPD have a somewhat higher verbal IQ and milder behavior problems.19,126,127 However, psychosis128 and autism spectrum disorders129,130 occur with significantly greater frequency among those with UPD. Individuals with the slightly larger, type 1 deletions (BP1–BP3) have been reported to have more compulsions and poorer adaptive behavior, intellectual ability, and academic achievement than those with type 2 deletions (BP2– BP3) deletions.131,132 Two other studies found much less clinically significant differences between individuals with these two deletion types.133,134
Diagnostic Testing
DNA methylation analysis is the most efficient way to start the genetic workup if PWS is suspected clinically ( Figure 5 and Table 3 ). The differential DNA methylation of several imprinted maternal and paternal loci in the 15q11.2-q13 region provides a powerful tool for assessing paternal-only, maternalonly, and normal (biparental) inheritance. DNA methylation analysis is the only technique that will diagnose PWS in all three molecular classes and differentiate PWS from AS in deletion cases,86,95 and a methylation analysis consistent with PWS is sufficient for clinical diagnosis (although not for genetic counseling purposes). It does not require parental DNA samples to differentiate the maternal and paternal alleles. Beginning in 1992, there have been three generations of DNA methylation clinical assays based on three distinct differentially methylated loci in the region.95,135,136 The most robust, and now most widely used, assay targets the 5′ CpG island of the SNURF-SNRPN (typically referred to as SNRPN) locus, and it will correctly diagnose PWS in more than 99% of cases.95,137 The promoter, exon 1, and intron 1 region of SNRPN are unmethylated on the paternally expressed allele and methylated on the maternally repressed allele. Normal individuals have both a methylated and an unmethylated allele, whereas individuals with PWS have only the maternally methylated allele. Methylation-specific multiplex-ligation probe amplification (MS-MLPA) can also determine the parental origin in this region and is discussed further below.

Algorithm for genetic testing for Prader-Willi syndrome (PWS). CMA, chromosomal microarray; FISH, fluorescence in situ hybridization; IC, imprinting center; MLPA, multiplex ligation probe amplification; UPD, uniparental disomy.
Although DNA methylation should be a first-line test for diagnosis, it cannot distinguish the molecular class (i.e., deletion, UPD, or ID). Therefore, once the diagnosis of PWS is established by DNA methylation analysis, determination of the molecular class is the next step. This determination is important for genetic counseling and genotype-phenotype correlation. It is usually most efficient to start by looking for a deletion as this is the most frequent cause of PWS ( Figure 4 ). Deletions of 15q11.2-q13 have traditionally been diagnosed with chromosomal analysis using FISH with the SNRPN probe. With the increasing use of CMA in clinical genetics, it is possible that arrays may replace FISH analysis for the identification of deletions in PWS (and AS). However, each technique has its advantages. CMA will precisely report the deletion size, which is anticipated to become increasingly important for genotype-phenotype correlations in the future (Driscoll and Kim, unpublished data). However, at this time, CMA is more expensive than chromosomal analysis with FISH testing in many commercial clinical laboratories, and arrays will not pick up the rare chromosomal rearrangements (translocations and inversions) involving proximal 15, which are detectable by simultaneous karyotype and FISH analysis and are important in recurrence risk determination. For genetic counseling purposes, a chromosomal analysis is advised in the proband to discern an interstitial de novo deletion from a balanced or unbalanced chromosomal rearrangement involving the 15q11.2 region. A CMA would also be indicated if an individual with PWS had a more severe phenotype than is typical, to discern if there was a larger deletion present or an additional chromosomal abnormality elsewhere in the genome.
Sometimes the first genetic test the individual with PWS has is chromosomal analysis with FISH or CMA rather than DNA methylation. If one of these techniques reveals a deletion in the PWS/AS region (i.e., 15q11.2-q13) and the child is younger than 2 years, it is still necessary to perform DNA methylation analysis as the deletions in PWS and AS are typically indistinguishable and AS can also present with hypotonia, feeding difficulties, and developmental delay in the neonatal period.91 In our clinical practice, the authors have independently been referred a number of infants who were “diagnosed” as PWS by a FISH deletion who, in fact, had AS. However, by 2–3 years of age, the clinical features of PWS and AS should become distinguishable.
If DNA methylation is positive for PWS (i.e., maternal only imprint), but no deletion is found, the next step is to distinguish between maternal UPD and an ID. This is accomplished by using DNA polymorphism analysis of chromosome 15 loci on the proband’s and parents’ DNA.138 If the family polymorphism study reveals that the proband has biparental inheritance of chromosome 15 loci (rather than maternal UPD), then the molecular class is presumed to be an ID. It is then important to determine whether the ID is due to an epimutation (low recurrence risk) or a small deletion in the IC, as in the later situation the recurrence risk can be as high as 50% if the father also has an IC deletion. Approximately 15% of individuals with PWS due to ID have an IC deletion and approximately 50% of these are familial mutations.88 Testing for an IC deletion should be done in a laboratory with experience in looking for IC deletions. This can be done by sequence analysis at the smallest region of overlap for the PWS IC, which is a region of approximately 4.3 kb,88,97 or by the recently developed MS-MLPA assay.
The commercially available MS-MLPA assay by MRC-Holland (Amsterdam, The Netherlands) combines both DNA methylation analysis and dosing analysis across the PWS region and has been shown to be very effective.139,140 The newest version as of this writing (ME028-B1) comes as a kit with 32 dosing probes specific for the 15q11.2 region and 14 probes outside the region (on chromosome 15 and other chromosomes), which serve as controls for copy number changes. In addition, five of the probes determine the DNA methylation status at differentially methylated sites in 15q11.2. The latest kit (B1) has particularly dense probe coverage for dosing and DNA methylation analysis in the PWS “critical region” between the PWS IC and SNORD116. Similar to CMA, MS-MLPA will not pick up chromosomal rearrangements (translocations and inversions) involving proximal 15, which are detectable by simultaneous karyotype and FISH analysis.
Some laboratories, particularly in Europe, now begin testing for PWS by using the MS-MLPA analysis (rather than single locus DNA methylation analysis at 5′SNRPN). The advantage of MS-MLPA over traditional DNA methylation analysis is that the MS-MLPA will investigate five distinct differentially methylated sites rather than just one locus and will give information on dosing in the 15q11.2 region. The MS-MLPA kit also will pick up IC deletions in PWS (and AS) and small deletions encompassing the SNORD116 gene cluster. If testing begins with MS-MLPA analysis, rather than single locus DNA methylation analysis, then the deletion status will be assessed at the same time as the DNA methylation status. However, neither single locus DNA methylation analysis nor MS-MLPA analysis will distinguish between the UPD and ID classes, and DNA polymorphism analysis of the parents and the proband is still necessary if no deletion is present and the DNA methylation is positive for PWS. Single-nucleotide polymorphism arrays can diagnose UPD in some cases but not all. However, DNA polymorphism analysis is still the gold standard test to use for diagnosing UPD.
Differential Diagnosis
Many disorders can mimic part of the PWS phenotype.
Craniopharyngioma and the results of its treatment and other types of damage to the hypothalamus show the greatest overlap with PWS, particularly if they occur at a young age.
Hyperphagic short stature is an acquired condition related to psychosocial stress that includes GH insufficiency, hyperphagia, and mild learning disabilities.141
Hypotonia in infancy is seen in many other conditions.142,143 Neonatal sepsis and central nervous system depression are common causes. Congenital myotonic dystrophy type 1 is characterized by hypotonia and severe generalized weakness at birth, often with respiratory insufficiency, developmental delay, and early death. A number of myopathies and neuropathies present as neonatal hypotonia, including some instances of spinal muscular atrophy. In these situations, poor respiratory effort may be present, a feature rarely seen in PWS. Molecular genetic testing, electromyography/nerve conduction velocity and/or muscle biopsy are often required to differentiate these conditions. A number of inborn errors of metabolism also may present with hypotonia with or without lethargy as their only finding in infancy. Pompe disease should particularly be considered. Newborn screening with tandem mass spectrometry will detect many inborn errors; metabolic disorder screening tests will detect others. Pompe disease must be confirmed by measurement of acid α-glucosidase enzyme activity or analysis of the GAA gene.
Several other genetic syndromes can present with neonatal hypotonia, including AS, which may have no other manifestations in the neonatal period. Affected individuals lack the characteristic sucking problems, hypogonadism, and facial appearance of PWS. Because AS can be caused by the same deletion as PWS, DNA methylation analysis is needed to distinguish them. Fragile X syndrome also includes hypotonia, which can be significant in the neonatal period.
In childhood, MECP2-related disorders (e.g., Rett syndrome) can present with hypotonia, obesity, and gynecomastia, as well as developmental disability. Affected individuals lack the characteristic history of sucking problems, hypogonadism, and facial appearance of PWS, and they often appear clinically normal in early infancy and have a progressive course.
Obesity and developmental delay/intellectual disability with or without hypogonadism can be seen in several genetic disorders. AS and Fragile X syndrome can both include obesity in a subset of individuals but not hypogonadism. Maternal UPD for chromosome 14 causes early motor and speech delay, excess weight, hypotonia, and can also include feeding problems, short stature, small hands and feet, and scoliosis.144 However, early puberty and joint laxity are typical. Albright hereditary osteodystrophy can include excess weight and developmental delays and also includes short stature and short metacarpals (especially 4th and 5th digits) but lacks hypotonia and has different characteristic facial appearance (round face). Bardet-Beidl syndrome includes truncal obesity, cognitive impairment, and male hypogonadotrophic hypogonadism but is distinguished by rodcone dystrophy, postaxial polydactyly, complex female genitourinary malformations, renal dysfunction, and a different facial phenotype from PWS. Cohen syndrome includes hypotonia, developmental disability (more severe than PWS), and obesity but has a different facial phenotype, microcephaly, progressive pigmentary retinopathy, severe myopia, and intermittent neutropenia. Borjeson-Forssman-Lehmann syndrome, seen in males, includes intellectual disability (severe), hypogonadism, marked obesity, infantile hypotonia and failure to thrive, and short stature. It can be distinguished by the severity of delays, the presence of nystagmus, and characteristic facial appearance, as well as genetic testing. Alstrom syndrome is characterized not only by early-onset obesity and developmental delay (approximately 50%) and sometimes male hypogonadotrophic hypogonadism but also by cone-rod dystrophy, progressive sensorineural hearing impairment, dilated cardiomyopathy (>60%), and the insulin resistance syndrome/type 2 diabetes mellitus associated with acanthosis nigricans, as well as urologic disorders of varying severity in females in their late teens and severe renal disease as a late finding.
A number of cytogenetic abnormalities result in overlap of manifestations with PWS, including deletion of 1p36, 2q37.3, 6q16.2, and 10q26 and duplication of 3p25.3.26.2 and Xq27.2-ter. Thus, CMA is an appropriate test when testing for PWS is negative or does not explain all the features.
Features similar to those of PWS in the presence of joint contractures suggest Urban-Roger, Camera, or Vasquez syndromes, all of which are rare.
Careful clinical evaluation by a medical geneticist or other trained diagnostician is useful to direct testing appropriately and may avoid the unnecessary expense of molecular testing for diagnoses that are less likely based on clinical findings.
Management
Management of the manifestations of PWS is age dependent and should include both addressing the consequences of the syndrome and anticipatory guidance. It is recommended that a team approach be used, if possible. Several other approaches to management have been published recently.4,34,145,146,147,148
Infancy
Special nipples or gavage feeding is usually needed to ensure adequate nutrition, as poor suck will result in failure to thrive if untreated. Growth measurements (height, weight, and head circumference) should be obtained and plotted at diagnosis and thereafter on a regular and frequent basis, at least every 2–3 months for the first year of life and as long as obtaining adequate calories is an issue. Those with prolonged failure to thrive despite adequate caloric intake should be assessed for hypothyroidism, which occurs in approximately 15%.63 Cryptorchidism should be sought and addressed with hormonal and/or surgical treatments in males. Many males with PWS can benefit from early treatment of hypogonadism (within the first 6 months of life) with either testosterone therapy or human chorionic gonadotropin (hCG) treatment to improve phallus size and assist with testicular descent into the scrotal sac.4 These therapies have the added benefit of improving muscle mass and strength. Clinical assessment for scoliosis should be done at diagnosis and annually, with follow-up X-rays should there be suspicion of its presence. GH replacement therapy to normalize height, increase lean body mass, mobility and activity level, and decrease fat mass is standard of care and can be started at any age.4,35,36,37,38,39,41,42,43 Studies thus far have indicated that no age is too early for GH treatment, and the sooner it is started the more benefit it has.42 It is recommended that a sleep study be performed before initiation of GH therapy to assess for and treat obstructive apnea.147 Developmental assessment and early intervention, to include physical therapy, should be focused on improving muscle strength and developmental milestone attainment. Referral for an ophthalmologic evaluation is indicated during the first year of life to assess for strabismus and visual acuity. Early diagnosis and multidisciplinary care have been shown to reduce hospital time and prevent early obesity.149
Childhood
Strict supervision of daily food intake based on height, weight, and BMI is required to ensure dequate energy requirements while limiting weight gain (keeping BMI <30). This should commence even before obesity is evident, to prevent its onset. Consultation with a dietician and close follow-up are usually necessary, and locking of the kitchen, refrigerator, and/or cupboards is often needed. The energy requirement of people with PWS, which rarely exceeds 1,000–1,200 Kcal/day,3 should be considered in planning daily food intake. Assessment of adequacy of vitamin and mineral intake by a dietician, and prescription of appropriate supplementation, is indicated, especially for calcium and vitamin D. Evaluation and treatment of sleep disturbance is recommended.147 Treatment depends on the cause and may include tonsillectomy and adenoidectomy and/or continuous positive airway pressure, as in the general population. GH treatment normalizes height, increases lean body mass, decreases fat mass, and increases mobility, which are beneficial to weight management.4,35,36,37,38,39,41,42,43 Dose recommendations in children are generally similar to those for individuals with isolated GH deficiency, i.e., approximately 1 mg/m2.42,147 Educational planning should be instigated, and speech therapy is often needed for articulation abnormalities and delays. An individual aide in the classroom is helpful in assuring attendance to task. Social skills training groups have been beneficial (personal observation). Firm limit setting is the first approach to limiting or treatment of behavioral problems; serotonin reuptake inhibitors are helpful for most individuals with more severe problems.150 Decreased saliva production can be addressed with products developed for the treatment of dry mouth, including special toothpastes, gels, mouthwash, and gum (personal observation). Issues of guardianship, wills, trusts, and advocacy should be investigated no later than adolescence.
Adolescence and adulthood
Management of hyperphagia and prevention of obesity are much the same as in children.46,48,49 The conventional adult dose of GH is 20–25% of the dose recommended in children. Replacement of sex hormones at puberty produces further development of secondary sexual characteristics (personal observation). However, systematic studies of sex hormone treatment in adolescents or adults with PWS are not available. If desired by the parents and child, treatment of hypogonadism usually commences at around age 11–12 years for females and age 12–13 for males. Females can be treated with low-dose estrogen therapy (usually by a transdermal patch to avoid interference with GH metabolism), with escalating doses for 2 years or until menarche, at which point they are transitioned to a combined estrogen-progesterone oral contraceptive pill or transdermal patch. The decision to treat hypogonadism in females with PWS is very much a personal decision for each family and typically depends on the maturity level, independence, and degree of obsessive-compulsive behaviors in the affected individual. Adolescent males with PWS can be treated with either a low-dose transdermal testosterone patch or gel with escalating doses every 3–6 months to allow the testosterone levels to get into the normal range for age or with hCG therapy. Treatment with hCG increases endogenous testosterone production, which increases testicular volume and lean body mass without causing the characteristic mood and aggressiveness problems which parents occasionally report with testosterone therapy.151
Consideration of sex education and contraception should occur, particularly in females with PWS, as pregnancy has been reported infrequently.27,28 Bone densitometry by DEXA every 1–2 years in adulthood is useful to evaluate for possible osteoporosis.147 Diabetes mellitus is usually avoided if obesity is avoided; calcium and vitamin D supplementation might aid in prevention of osteoporosis. Scoliosis should be sought at regular examinations throughout life. Routine monitoring of weight and BMI is important to ensure appropriateness of exercise program and diet. No currently available medication or surgical approach has been shown to aid in controlling hyperphagia. Daily muscle training increases physical activity and lean body mass.152 Psychosis is reported to respond well to selective serotonin reuptake inhibitors but not to mood stabilizers.150 A group home for individuals with PWS that incorporates regulation of behavior and weight and ensures adequate physical activity may prevent morbid obesity (personal observation).
Genetic Counseling
Knowing the specific genetic etiology in individuals with PWS is essential for the appropriate genetic counseling of affected families ( Table 4 ). The majority of families have a recurrence risk less than 1%. However, certain etiologies have a recurrence risk as high as 50%, and a scenario with a risk of almost 100% is very unlikely but theoretically possible (i.e., a mother with a 15/15 Robertsonian translocation).
Nearly all 15q11.2-q13 deletions are de novo interstitial deletions (Ia) with a very low recurrence risk (<1%). A chromosome analysis with FISH should be performed in individuals with a deletion as, on rare occasions, the deletion is the result of a chromosomal rearrangement (Ib). This could have occurred de novo in the proband’s father’s gamete, or the father may carry a balanced rearrangement. There have been several reports of a balanced rearrangement (translocation or inversion) in the father resulting in a deletion of the 15q11.2 region.153,154 In these cases, there is a theoretical recurrence risk as high as 25–50%. On rare occasions, a second chromosomal anomaly is noted in addition to the 15q11.2 deletion, such as Klinefelter syndrome.155,156,157 Counseling of the family regarding the clinical prognosis of the proband is thus influenced by the presence of additional chromosomal anomalies.
A parental chromosomal rearrangement is not typically obvious using only the proband’s chromosome and FISH analyses. For example, a paternal paracentric inversion within or including the 15q11.2-q13 region with unequal crossing over in paternal meiosis could result in a deletion in the offspring. A normal brother and sister with a paracentric inversion involving the 15q11.2-q13 region has been reported.158 The brother had a child with PWS and the sister had a child with AS. Therefore, fathers of children with deletion should be offered chromosomal and FISH analyses for the 15q11.2-q13 region. FISH analysis of individuals with PWS and their fathers should include at a minimum the simultaneous use of a chromosome 15 p arm or centromeric probe (e.g., D15Z1), a critical region probe (e.g., SNRPN) and a distal control probe (e.g., PML at 15q22). The use of a 15p or centromeric probe is crucial in diagnosing a cryptic translocation, particularly between two acrocentric chromosomes. Furthermore, it would be ideal for the examination of the father’s chromosomes 15 to include the use of two differentially colored critical region probes (e.g., SNRPN and D15S10) to evaluate for the possibility of an inversion. It is technically much harder to use two (rather than only one) critical region probes due to the difficulty in getting sufficient separation of the probes in condensed chromosomes. However, adequate separation, although technically difficult, could be possible in interphase, with orientation provided by the short arm/centromeric and distal 15 probes.
Maternal UPD 15 is typically de novo (IIa), with a recurrence <1% except if a Robertsonian translocation is present in either parent, so a chromosomal analysis is indicated in the proband. If this is normal, then the father of the child should be offered a chromosomal analysis to ensure that he does not have a Robertsonian translocation. We presume that the mother does not have a Robertsonian translocation as the two maternal chromosomes 15 are normal in the proband. However, we cannot rule out the possibility that the father has a Robertsonian translocation involving chromosome 15, which led to aberrant segregation at meiosis I and resulted in a sperm that was nullisomic for 15. This, combined with monosomy rescue to disomy, would result in an embryo with maternal UPD 15.
Rarely, a small marker chromosome is also present in a proband with maternal UPD 15.121 In these instances, it is important to examine both parents’ karyotype as it seems that these small marker chromosomes may increase the risk for nondisjunction and UPD.122
People with PWS due to an ID should be tested for an IC deletion by a laboratory experienced in detecting them. The majority (approximately 85%) of those with an ID have a de novo epigenetic mutation (IIIb) and the recurrence risk is <1% for this group. However, approximately 15% of those with an ID have it on the basis of a microdeletion in the IC (IIIa). In approximately half of these individuals, the IC deletion is familial and the recurrence risk is 50% for these families. Therefore, fathers of children with an IC deletion should have DNA methylation and dosing analysis (or sequence analysis) to determine whether they carry the IC deletion.
Offspring of a proband
With rare exception, individuals with PWS do not reproduce. However, there are two reported27,28 and two as-yet unreported (Cassidy and Vats, unpublished data) women with genetically confirmed PWS who have had a child. No genetically confirmed males with PWS have been known to have fathered a child. The risk to the child of an affected individual depends on the etiology of the PWS and the sex of the affected individual. If the proband has PWS as the result of a deletion, the offspring have a 50% risk of having AS if the proband is female28 and PWS if the proband is male (never reported). There is a single report of a female with PWS caused by UPD having a normal child.27
Prenatal testing
Families who have children with PWS should be aware that prenatal diagnosis for PWS is available and will not be typically diagnosed in a standard prenatal chromosome analysis. Germ cell mosaicism in the father is a rare but distinct possibility and has been observed in cases of 15q11.2 deletions159 and IC deletions.123,160 In addition, recurrent meiotic nondisjunction of maternal chromosome 15 has been observed.161 FISH, array comparative genomic hybridization, DNA methylation analysis, MS-MLPA, and DNA polymorphism studies for UPD have been validated in prenatal diagnosis, but only DNA methylation analysis (including MS-MLPA) at the 5′ SNRPN locus will identify the IDs.162,163,164 It should be noted that only a few clinical laboratories have the experience to use DNA methylation analysis in prenatal diagnosis and these laboratories typically prefer to use amniocytes (versus chorionic villi) for analysis because of the known hypomethylation of tissue derived from the placenta.163,165
Disclosure
S.S. is an employee of Laboratory Corporation of America, which does genetic testing.
References
Butler MG, Sturich J, Myers SE, Gold JA, Kimonis V, Driscoll DJ . Is gestation in Prader-Willi syndrome affected by the genetic subtype? J Assist Reprod Genet 2009;26:461–466.
Butler MG, Sturich J, Lee J, et al. Growth standards of infants with Prader-Willi syndrome. Pediatrics 2011;127:687–695.
Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A 2011;155A:1040–1049.
McCandless SE ; Committee on Genetics. Clinical report—health supervision for children with Prader-Willi syndrome. Pediatrics 2011;127:195–204.
Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H . Academic underachievement by people with Prader-Willi syndrome. J Intellect Disabil Res 2004;48(Pt 2):188–200.
Butler MG, Theodoro MF, Bittel DC, Donnelly JE . Energy expenditure and physical activity in Prader-Willi syndrome: comparison with obese subjects. Am J Med Genet A 2007;143:449–459.
Goldstone AP, Thomas EL, Brynes AE, et al. Visceral adipose tissue and metabolic complications of obesity are reduced in Prader-Willi syndrome female adults: evidence for novel influences on body fat distribution. J Clin Endocrinol Metab 2001;86:4330–4338.
Cummings DE, Clement K, Purnell JQ, et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med 2002;8:643–644.
DelParigi A, Tschöp M, Heiman ML, et al. High circulating ghrelin: a potential cause for hyperphagia and obesity in prader-willi syndrome. J Clin Endocrinol Metab 2002;87:5461–5464.
Haqq AM, Farooqi IS, O’Rahilly S, et al. Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi syndrome. J Clin Endocrinol Metab 2003;88:174–178.
Goldstone AP, Thomas EL, Brynes AE, et al. Elevated fasting plasma ghrelin in prader-willi syndrome adults is not solely explained by their reduced visceral adiposity and insulin resistance. J Clin Endocrinol Metab 2004;89:1718–1726.
Goldstone AP, Patterson M, Kalingag N, et al. Fasting and postprandial hyperghrelinemia in Prader-Willi syndrome is partially explained by hypoinsulinemia, and is not due to peptide YY3-36 deficiency or seen in hypothalamic obesity due to craniopharyngioma. J Clin Endocrinol Metab 2005;90:2681–2690.
Scerif M, Goldstone AP, Korbonits M . Ghrelin in obesity and endocrine diseases. Mol Cell Endocrinol 2011;340:15–25.
Erdie-Lalena CR, Holm VA, Kelly PC, Frayo RS, Cummings DE . Ghrelin levels in young children with Prader-Willi syndrome. J Pediatr 2006;149:199–204.
Feigerlová E, Diene G, Conte-Auriol F, et al. Hyperghrelinemia precedes obesity in Prader-Willi syndrome. J Clin Endocrinol Metab 2008;93:2800–2805.
Haqq AM, Stadler DD, Rosenfeld RG, et al. Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi syndrome. J Clin Endocrinol Metab 2003;88:3573–3576.
Tan TM, Vanderpump M, Khoo B, Patterson M, Ghatei MA, Goldstone AP . Somatostatin infusion lowers plasma ghrelin without reducing appetite in adults with Prader-Willi syndrome. J Clin Endocrinol Metab 2004;89:4162–4165.
De Waele K, Ishkanian SL, Bogarin R, et al. Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader-Willi syndrome. Eur J Endocrinol 2008;159:381–388.
Dykens EM, Cassidy SB, King BH . Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am J Ment Retard 1999;104:67–77.
Descheemaeker MJ, Govers V, Vermeulen P, Fryns JP . Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A 2006;140:1136–1142.
Wigren M, Hansen S . ADHD symptoms and insistence on sameness in Prader-Willi syndrome. J Intellect Disabil Res 2005;49(Pt 6):449–456.
Steinhausen HC, Eiholzer U, Hauffa BP, Malin Z . Behavioural and emotional disturbances in people with Prader-Willi Syndrome. J Intellect Disabil Res 2004;48:47–52.
Dykens EM . Maladaptive and compulsive behavior in Prader-Willi syndrome: new insights from older adults. Am J Ment Retard 2004;109:142–153.
Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D . Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet 2002;359:135–136.
Clarke DJ, Boer H, Whittington J, Holland A, Butler J, Webb T . Prader-Willi syndrome, compulsive and ritualistic behaviours: the first population-based survey. Br J Psychiatry 2002;180:358–362.
Vogels A, De Hert M, Descheemaeker MJ, et al. Psychotic disorders in Prader-Willi syndrome. Am J Med Genet A 2004;127A:238–243.
Akefeldt A, Törnhage CJ, Gillberg C . ‘A woman with Prader-Willi syndrome gives birth to a healthy baby girl’. Dev Med Child Neurol 1999;41:789–790.
Schulze A, Mogensen H, Hamborg-Petersen B, Graem N, Ostergaard JR, Brøndum-Nielsen K . Fertility in Prader-Willi syndrome: a case report with Angelman syndrome in the offspring. Acta Paediatr 2001;90:455–459.
Crinò A, Schiaffini R, Ciampalini P, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr 2003;162:327–333.
Hirsch HJ, Eldar-Geva T, Benarroch F, Rubinstein O, Gross-Tsur V . Primary testicular dysfunction is a major contributor to abnormal pubertal development in males with Prader-Willi syndrome. J Clin Endocrinol Metab 2009;94:2262–2268.
Eldar-Geva T, Hirsch HJ, Rabinowitz R, Benarroch F, Rubinstein O, Gross-Tsur V . Primary ovarian dysfunction contributes to the hypogonadism in women with Prader-Willi Syndrome. Horm Res 2009;72:153–159.
Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O, Gross-Tsur V . Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur J Endocrinol 2010;162:377–384.
Burman P, Ritzén EM, Lindgren AC . Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr Rev 2001;22:787–799.
Cassidy SB, McCandless SM . Prader-Willi syndrome. In: Cassidy SB, Allanson JE (eds). Management of Genetic Syndromes, 3rd edn. John Wiley & Sons: New York, 2010:625–50.
Myers SE, Carrel AL, Whitman BY, Allen DB . Sustained benefit after 2 years of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome. J Pediatr 2000;137:42–49.
Eiholzer U, Schlumpf M, Nordmann Y, l’Allemand D . Early manifestations of Prader-Willi syndrome: influence of growth hormone. J Pediatr Endocrinol Metab 2001;14(suppl 6):1441–1444.
Eiholzer U, L’allemand D, Schlumpf M, Rousson V, Gasser T, Fusch C . Growth hormone and body composition in children younger than 2 years with Prader-Willi syndrome. J Pediatr 2004;144:753–758.
Whitman B, Carrel A, Bekx T, Weber C, Allen D, Myers S . Growth hormone improves body composition and motor development in infants with Prader-Willi syndrome after six months. J Pediatr Endocrinol Metab 2004;17:591–600.
Festen DA, de Lind van Wijngaarden R, van Eekelen M, et al. Randomized controlled GH trial: effects on anthropometry, body composition and body proportions in a large group of children with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2008;69:443–451.
de Lind van Wijngaarden RF, de Klerk LW, Festen DA, Duivenvoorden HJ, Otten BJ, Hokken-Koelega AC . Randomized controlled trial to investigate the effects of growth hormone treatment on scoliosis in children with Prader-Willi syndrome. J Clin Endocrinol Metab 2009;94:1274–1280.
Nyunt O, Harris M, Hughes I, Huynh T, Davies PS, Cotterill AM . Benefit of early commencement of growth hormone therapy in children with Prader-Willi syndrome. J Pediatr Endocrinol Metab 2009;22:1151–1158.
Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB . Long-term growth hormone therapy changes the natural history of body composition and motor function in children with prader-willi syndrome. J Clin Endocrinol Metab 2010;95:1131–1136.
Colmenares A, Pinto G, Taupin P, et al. Effects on growth and metabolism of growth hormone treatment for 3 years in 36 children with Prader-Willi syndrome. Horm Res Paediatr 2011;75:123–130.
Whitman BY, Myers S, Carrel A, Allen D . The behavioral impact of growth hormone treatment for children and adolescents with Prader-Willi syndrome: a 2-year, controlled study. Pediatrics 2002;109:E35.
Grugni G, Marzullo P, Ragusa L, et al. Impairment of GH responsiveness to combined GH-releasing hormone and arginine administration in adult patients with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2006;65:492–499.
Höybye C . Five-years growth hormone (GH) treatment in adults with Prader-Willi syndrome. Acta Paediatr 2007;96:410–413.
Grugni G, Giardino D, Crinò A, et al. Growth hormone secretion among adult patients with Prader-Willi syndrome due to different genetic subtypes. J Endocrinol Invest 2011;34:493–497.
Mogul HR, Lee PD, Whitman BY, et al. Growth hormone treatment of adults with Prader-Willi syndrome and growth hormone deficiency improves lean body mass, fractional body fat, and serum triiodothyronine without glucose impairment: results from the United States multicenter trial. J Clin Endocrinol Metab 2008;93:1238–1245.
Sode-Carlsen R, Farholt S, Rabben KF, et al. One year of growth hormone treatment in adults with Prader-Willi syndrome improves body composition: results from a randomized, placebo-controlled study. J Clin Endocrinol Metab 2010;95:4943–4950.
Eiholzer U, Nordmann Y, L’Allemand D . Fatal outcome of sleep apnoea in PWS during the initial phase of growth hormone treatment. A case report. Horm Res 2002;58(suppl 3):24–26.
Nordmann Y, Eiholzer U, l’Allemand D, Mirjanic S, Markwalder C . Sudden death of an infant with Prader-Willi syndrome–not a unique case? Biol Neonate 2002;82:139–141.
Van Vliet G, Deal CL, Crock PA, Robitaille Y, Oligny LL . Sudden death in growth hormone-treated children with Prader-Willi syndrome. J Pediatr 2004;144:129–131.
Eiholzer U . Deaths in children with Prader-Willi syndrome. A contribution to the debate about the safety of growth hormone treatment in children with PWS. Horm Res 2005;63:33–39.
Sacco M, Di Giorgio G . Sudden death in Prader-Willi syndrome during growth hormone therapy. Horm Res 2005;63:29–32.
Bakker B, Maneatis T, Lippe B . Sudden death in Prader-Willi syndrome: brief review of five additional cases. Concerning the article by U. Eiholzer et al.: Deaths in children with Prader-Willi syndrome. A contribution to the debate about the safety of growth hormone treatment in children with PWS (Horm Res 2005;63:33–39). Horm Res 2007;67:203–4.
Craig ME, Cowell CT, Larsson P, et al. Growth hormone treatment and adverse events in Prader-Willi syndrome: data from KIGS (the Pfizer International Growth Database). Clin Endocrinol (Oxf) 2006;65:178–185.
Nagai T, Obata K, Tonoki H, et al. Cause of sudden, unexpected death of Prader-Willi syndrome patients with or without growth hormone treatment. Am J Med Genet A 2005;136:45–48.
Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H . Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet 2001;38:792–798.
de Lind van Wijngaarden RF, Otten BJ, Festen DA, et al. High prevalence of central adrenal insufficiency in patients with Prader-Willi syndrome. J Clin Endocrinol Metab 2008;93:1649–1654.
Scaroni C, Ceccato F, Rizzati S, Mantero F . Concomitant therapies (glucocorticoids and sex hormones) in adult patients with growth hormone deficiency. J Endocrinol Invest 2008;31(suppl 9):61–65.
Nyunt O, Cotterill AM, Archbold SM, et al. Normal cortisol response on low-dose synacthen (1 microg) test in children with Prader Willi syndrome. J Clin Endocrinol Metab 2010;95:E464–E467.
Farholt S, Sode-Carlsen R, Christiansen JS, Østergaard JR, Høybye C . Normal cortisol response to high-dose synacthen and insulin tolerance test in children and adults with Prader-Willi syndrome. J Clin Endocrinol Metab 2011;96:E173–E180.
Miller JL, Goldstone AP, Couch JA, et al. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am J Med Genet A 2008;146A:570–577.
Diene G, Mimoun E, Feigerlova E, et al.; French Reference Centre for PWS. Endocrine disorders in children with Prader-Willi syndrome–data from 142 children of the French database. Horm Res Paediatr 2010;74:121–128.
Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T . Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol 2002;44:248–255.
Festen DA, de Weerd AW, van den Bossche RA, Joosten K, Hoeve H, Hokken-Koelega AC . Sleep-related breathing disorders in prepubertal children with Prader-Willi syndrome and effects of growth hormone treatment. J Clin Endocrinol Metab 2006;91:4911–4915.
Priano L, Grugni G, Miscio G, et al. Sleep cycling alternating pattern (CAP) expression is associated with hypersomnia and GH secretory pattern in Prader-Willi syndrome. Sleep Med 2006;7:627–633.
Dauvilliers Y, Baumann CR, Carlander B, et al. CSF hypocretin-1 levels in narcolepsy, Kleine-Levin syndrome, and other hypersomnias and neurological conditions. J Neurol Neurosurg Psychiatr 2003;74:1667–1673.
Nevsimalova S, Vankova J, Stepanova I, Seemanova E, Mignot E, Nishino S . Hypocretin deficiency in Prader-Willi syndrome. Eur J Neurol 2005;12:70–72.
Bruni O, Verrillo E, Novelli L, Ferri R . Prader-Willi syndrome: sorting out the relationships between obesity, hypersomnia, and sleep apnea. Curr Opin Pulm Med 2010;16:568–573.
Hayashi M, Miyata R, Tanuma N . Decrease in acetylcholinergic neurons in the pedunculopontine tegmental nucleus in a patient with Prader-Willi syndrome. Neuropathology 2011;31:280–285.
West LA, Ballock RT . High incidence of hip dysplasia but not slipped capital femoral epiphysis in patients with Prader-Willi syndrome. J Pediatr Orthop 2004;24:565–567.
Shim JS, Lee SH, Seo SW, Koo KH, Jin DK . The musculoskeletal manifestations of Prader-Willi syndrome. J Pediatr Orthop 2010;30:390–395.
Miller JL, Couch JA, Schmalfuss I, He G, Liu Y, Driscoll DJ . Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in Prader-Willi syndrome. Am J Med Genet A 2007;143:476–483.
Miller J, Kranzler J, Liu Y, et al. Neurocognitive findings in Prader-Willi syndrome and early-onset morbid obesity. J Pediatr 2006;149:192–198.
Iughetti L, Bosio L, Corrias A, et al. Pituitary height and neuroradiological alterations in patients with Prader-Labhart-Willi syndrome. Eur J Pediatr 2008;167:701–702.
Einfeld SL, Kavanagh SJ, Smith A, Evans EJ, Tonge BJ, Taffe J . Mortality in Prader-Willi syndrome. Am J Ment Retard 2006;111:193–198.
Schrander-Stumpel CT, Curfs LM, Sastrowijoto P, Cassidy SB, Schrander JJ, Fryns JP . Prader-Willi syndrome: causes of death in an international series of 27 cases. Am J Med Genet A 2004;124A:333–338.
Stevenson DA, Anaya TM, Clayton-Smith J, et al. Unexpected death and critical illness in Prader-Willi syndrome: report of ten individuals. Am J Med Genet A 2004;124A:158–164.
Tauber M, Diene G, Molinas C, Hébert M . Review of 64 cases of death in children with Prader-Willi syndrome (PWS). Am J Med Genet A 2008;146:881–887.
Wharton RH, Wang T, Graeme-Cook F, Briggs S, Cole RE . Acute idiopathic gastric dilation with gastric necrosis in individuals with Prader-Willi syndrome. Am J Med Genet 1997;73:437–441.
Stevenson DA, Heinemann J, Angulo M, et al. Gastric rupture and necrosis in Prader-Willi syndrome. J Pediatr Gastroenterol Nutr 2007;45:272–274.
Stevenson DA, Heinemann J, Angulo M, et al. Deaths due to choking in Prader-Willi syndrome. Am J Med Genet A 2007;143:484–487.
Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 1993;91:398–402.
Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB . The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001;108:E92.
Glenn CC, Driscoll DJ, Yang TP, Nicholls RD . Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod 1997;3:321–332.
Bittel DC, Butler MG . Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med 2005;7:1–20.
Horsthemke B, Buiting K . Genomic imprinting and imprinting defects in humans. Adv Genet 2008;61:225–246.
Horsthemke B . Mechanisms of imprint dysregulation. Am J Med Genet C Semin Med Genet 2010;154C:321–328.
Lossie AC, Whitney MM, Amidon D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet 2001;38:834–845.
Williams CA, Driscoll DJ, Dagli AI . Clinical and genetic aspects of Angelman syndrome. Genet Med 2010;12:385–395.
Christian SL, Robinson WP, Huang B, et al. Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet 1995;57:40–48.
Amos-Landgraf JM, Ji Y, Gottlieb W, et al. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 1999;65:370–386.
Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet 2003;73:898–925.
Glenn CC, Saitoh S, Jong MT, et al. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet 1996;58:335–346.
Gray TA, Saitoh S, Nicholls RD . An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc Natl Acad Sci USA 1999;96:5616–5621.
Ohta T, Gray TA, Rogan PK, et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet 1999;64:397–413.
Cavaillé J, Buiting K, Kiefmann M, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci USA 2000;97:14311–14316.
Runte M, Hüttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K . The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet 2001;10:2687–2700.
Chamberlain SJ, Brannan CI . The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics 2001;73:316–322.
Kishore S, Stamm S . Regulation of alternative splicing by snoRNAs. Cold Spring Harb Symp Quant Biol 2006;71:329–334.
Buiting K . Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 2010;154C:365–376.
Runte M, Varon R, Horn D, Horsthemke B, Buiting K . Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader-Willi syndrome. Hum Genet 2005;116:228–230.
Sahoo T, del Gaudio D, German JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 2008;40:719–721.
de Smith AJ, Purmann C, Walters RG, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet 2009;18:3257–3265.
Duker AL, Ballif BC, Bawle EV, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 2010;18:1196–1201.
Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD . Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med 1981;304:325–329.
Butler MG, Palmer CG . Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet 1983;1:1285–1286.
Robinson WP, Dutly F, Nicholls RD, et al. The mechanisms involved in formation of deletions and duplications of 15q11-q13. J Med Genet 1998;35:130–136.
Boyar FZ, Whitney MM, Lossie AC, et al. A family with a grand-maternally derived interstitial duplication of proximal 15q. Clin Genet 2001;60:421–430.
Maggouta F, Roberts SE, Dennis NR, Veltman MW, Crolla JA . A supernumerary marker chromosome 15 tetrasomic for the Prader-Willi/Angelman syndrome critical region in a patient with a severe phenotype. J Med Genet 2003;40:e84.
Depienne C, Moreno-De-Luca D, Heron D, et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry 2009;66:349–359.
Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M . Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 1989;342:281–285.
Yamazawa K, Ogata T, Ferguson-Smith AC . Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet 2010;154C:329–334.
Gillessen-Kaesbach G, Robinson W, Lohmann D, Kaya-Westerloh S, Passarge E, Horsthemke B . Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum Genet 1995;96:638–643.
Cassidy SB, Forsythe M, Heeger S, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet 1997;68:433–440.
Cassidy SB, Lai LW, Erickson RP, et al. Trisomy 15 with loss of the paternal 15 as a cause of Prader-Willi syndrome due to maternal disomy. Am J Hum Genet 1992;51:701–708.
Purvis-Smith SG, Saville T, Manass S, et al. Uniparental disomy 15 resulting from “correction” of an initial trisomy 15. Am J Hum Genet 1992;50:1348–1350.
Robinson WP, Barrett IJ, Bernard L, et al. Meiotic origin of trisomy in confined placental mosaicism is correlated with presence of fetal uniparental disomy, high levels of trisomy in trophoblast, and increased risk of fetal intrauterine growth restriction. Am J Hum Genet 1997;60:917–927.
EUCROMIC. Trisomy 15 CPM: probable origins, pregnancy outcome and risk of fetal UPD. European Collaborative Research on Mosaicism in CVS. Prenat Diagn 1999;19:29–35.
Liehr T, Brude E, Gillessen-Kaesbach G, et al. Prader-Willi syndrome with a karyotype 47,XY,+min(15)(pter->q11.1:) and maternal UPD 15–case report plus review of similar cases. Eur J Med Genet 2005;48:175–181.
Kotzot D . Review and meta-analysis of systematic searches for uniparental disomy (UPD) other than UPD 15. Am J Med Genet 2002;111:366–375.
Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B . Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet 2003;72:571–577.
Butler MG . Hypopigmentation: a common feature of Prader-Labhart-Willi syndrome. Am J Hum Genet 1989;45:140–146.
Dykens EM . Are jigsaw puzzle skills ‘spared’ in persons with Prader-Willi syndrome? J Child Psychol Psychiatry 2002;43:343–352.
Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG . Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res 2000;44(Pt 1):25–30.
Torrado M, Araoz V, Baialardo E, et al. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A 2007;143:460–468.
Holland AJ, Whittington JE, Butler J, Webb T, Boer H, Clarke D . Behavioural phenotypes associated with specific genetic disorders: evidence from a population-based study of people with Prader-Willi syndrome. Psychol Med 2003;33:141–153.
Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF . Prader-Willi syndrome–a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry 2004;13:42–50.
Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H . Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. J Intellect Disabil Res 2004;48(Pt 2):172–187.
Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T . Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004;113(3 Pt 1):565–573.
Hartley SL, Maclean WE Jr, Butler MG, Zarcone J, Thompson T . Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am J Med Genet A 2005;136:140–145.
Milner KM, Craig EE, Thompson RJ, et al. Prader-Willi syndrome: intellectual abilities and behavioural features by genetic subtype. J Child Psychol Psychiatry 2005;46:1089–1096.
Varela MC, Kok F, Setian N, Kim CA, Koiffmann CP . Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: study of 75 patients. Clin Genet 2005;67:47–52.
Driscoll DJ, Waters MF, Williams CA, et al. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics 1992;13:917–924.
Dittrich B, Buiting K, Gross S, Horsthemke B . Characterization of a methylation imprint in the Prader-Willi syndrome chromosome region. Hum Mol Genet 1993;2:1995–1999.
Kubota T, Das S, Christian SL, Baylin SB, Herman JG, Ledbetter DH . Methylation-specific PCR simplifies imprinting analysis. Nat Genet 1997;16:16–17.
Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB . American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet Med 2001;3:206–211.
Procter M, Chou LS, Tang W, Jama M, Mao R . Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem 2006;52:1276–1283.
Bittel DC, Kibiryeva N, Sell SM, Strong TV, Butler MG . Whole genome microarray analysis of gene expression in Prader-Willi syndrome. Am J Med Genet A 2007;143:430–442.
Gilmour J, Skuse D, Pembrey M . Hyperphagic short stature and Prader–Willi syndrome: a comparison of behavioural phenotypes, genotypes and indices of stress. Br J Psychiatry 2001;179:129–137.
Miller SP, Riley P, Shevell MI . The neonatal presentation of Prader-Willi syndrome revisited. J Pediatr 1999;134:226–228.
Richer LP, Shevell MI, Miller SP . Diagnostic profile of neonatal hypotonia: an 11-year study. Pediatr Neurol 2001;25:32–37.
Cox H, Bullman H, Temple IK . Maternal UPD(14) in the patient with a normal karyotype: clinical report and a systematic search for cases in samples sent for testing for Prader-Willi syndrome. Am J Med Genet A 2004;127A:21–25.
Eiholzer U, Whitman BY . A comprehensive team approach to the management of patients with Prader-Willi syndrome. J Pediatr Endocrinol Metab 2004;17:1153–1175.
Butler MG, Lee PD, Whitman BY . Management of Prader-Willi Syndrome, 3rd edn. Springer: New York, 2006.
Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M ; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab 2008;93:4183–4197.
Cassidy SB, Driscoll DJ . Prader-Willi syndrome. Eur J Hum Genet 2009;17:3–13.
Bacheré N, Diene G, Delagnes V, Molinas C, Moulin P, Tauber M . Early diagnosis and multidisciplinary care reduce the hospitalization time and duration of tube feeding and prevent early obesity in PWS infants. Horm Res 2008;69:45–52.
Soni S, Whittington J, Holland AJ, et al. The course and outcome of psychiatric illness in people with Prader-Willi syndrome: implications for management and treatment. J Intellect Disabil Res 2007;51(Pt 1):32–42.
Eiholzer U, Grieser J, Schlumpf M, l’Allemand D . Clinical effects of treatment for hypogonadism in male adolescents with Prader-Labhart-Willi syndrome. Horm Res 2007;68:178–184.
Schlumpf M, Eiholzer U, Gygax M, Schmid S, van der Sluis I, l’Allemand D . A daily comprehensive muscle training programme increases lean mass and spontaneous activity in children with Prader-Willi syndrome after 6 months. J Pediatr Endocrinol Metab 2006;19:65–74.
Horsthemke B, Maat-Kievit A, Sleegers E, et al. Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet 1996;33:848–851.
Flori E, Biancalana V, Girard-Lemaire F, et al. Difficulties of genetic counseling and prenatal diagnosis in a consanguineous couple segregating for the same translocation (14;15) (q11;q13) and at risk for Prader-Willi and Angelman syndromes. Eur J Hum Genet 2004;12:181–186.
Butler MG, Hedges LK, Rogan PK, Seip JR, Cassidy SB, Moeschler JB . Klinefelter and trisomy X syndromes in patients with Prader-Willi syndrome and uniparental maternal disomy of chromosome 15–a coincidence? Am J Med Genet 1997;72:111–114.
Rego A, Coll MD, Regal M, Guitart M, Escudero T, García-Mayor RV . A case with 47,XXY,del(15)(q11;q13) karyotype associated with Prader-Willi phenotype. Horm Res 1997;48:44–46.
Verhoeven WM, de Vries BB, Duffels SJ, Egger JI, Noordam C, Tuinier S . Klinefelter’s syndrome and Prader-Willi syndrome: a rare combination. Psychopathology 2007;40:356–360.
Clayton-Smith J, Driscoll DJ, Waters MF, et al. Difference in methylation patterns within the D15S9 region of chromosome 15q11-13 in first cousins with Angelman syndrome and Prader-Willi syndrome. Am J Med Genet 1993;47:683–686.
Fernaández-Novoa MC, Vargas MT, Vizmanos JL, et al. Prader-Willi syndrome large deletion on two brothers. Is this the exception that confirm the rule? Rev Neurol 2001;32:935–938.
Wey E, Bartholdi D, Riegel M, et al. Mosaic imprinting defect in a patient with an almost typical expression of the Prader-Willi syndrome. Eur J Hum Genet 2005;13:273–277.
Harpey JP, Heron D, Prudent M, et al. Recurrent meiotic nondisjunction of maternal chromosome 15 in a sibship. Am J Med Genet 1998;76:103–104.
Kubota T, Sutcliffe JS, Aradhya S, et al. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet 1996;66:77–80.
Glenn CC, Deng G, Michaelis RC, et al. DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diagn 2000;20:300–306.
Ramsden SC, Clayton-Smith J, Birch R, Buiting K . Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet 2010;11:70.
Driscoll DJ, Migeon BR . Sex difference in methylation of single-copy genes in human meiotic germ cells: implications for X chromosome inactivation, parental imprinting, and origin of CpG mutations. Somat Cell Mol Genet 1990;16:267–282.
Acknowledgements
The authors thank the many individuals with Prader-Willi syndrome and their families who have contributed to our clinical experience, research, and educational efforts. The authors also thank Fred Kweh for his expert help with the figures.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cassidy, S., Schwartz, S., Miller, J. et al. Prader-Willi syndrome. Genet Med 14, 10–26 (2012). https://doi.org/10.1038/gim.0b013e31822bead0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.0b013e31822bead0
Keywords
This article is cited by
-
Caregiver-Implemented Behavior Analytic Treatment Package for Skin Picking in PWS: A Pilot Study
Advances in Neurodevelopmental Disorders (2024)
-
The outcomes of growth hormone therapy in the obstructive sleep apnea parameters of Prader–Willi syndrome patients: a systematic review
European Archives of Oto-Rhino-Laryngology (2024)
-
Systemic immune profile in Prader-Willi syndrome: elevated matrix metalloproteinase and myeloperoxidase and reduced macrophage inhibitory factor
Orphanet Journal of Rare Diseases (2023)
-
Aberrant brain intra- and internetwork functional connectivity in children with Prader-Willi syndrome
Neuroradiology (2023)
-
Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD): a collaborative review of the current understanding
Clinical Autonomic Research (2023)
