
Abstract
Alectinib, an inhibitor of anaplastic lymphoma kinase (ALK), was approved by the Food and Drug Administration (FDA) for the treatment of patients with ALK-positive non-small cell lung cancer (NSCLC). Here we investigated the reversal effect of alectinib on multidrug resistance (MDR) induced by ATP-binding cassette (ABC) transporters, which is the primary cause of chemotherapy failure. We provide the first evidence that alectinib increases the sensitivity of ABCB1- and ABCG2-overexpressing cells to chemotherapeutic agents in vitro and in vivo. Mechanistically, alectinib increased the intracellular accumulation of ABCB1/ABCG2 substrates such as doxorubicin (DOX) and Rhodamine 123 (Rho 123) by inhibiting the efflux function of the transporters in ABCB1- or ABCG2-overexpressing cells but not in their parental sensitive cells. Furthermore, alectinib stimulated ATPase activity and competed with substrates of ABCB1 or ABCG2 and competed with [125I] iodoarylazidoprazosin (IAAP) photolabeling bound to ABCB1 or ABCG2 but neither altered the expression and localization of ABCB1 or ABCG2 nor the phosphorylation levels of AKT and ERK. Alectinib also enhanced the cytotoxicity of DOX and the intracellular accumulation of Rho 123 in ABCB1-overexpressing primary leukemia cells. These findings suggest that alectinib combined with traditional chemotherapy may be beneficial to patients with ABCB1- or ABCG2-mediated MDR.
Similar content being viewed by others


Introduction
Chemotherapy is the mainstay of treatment for many human cancers. Unfortunately, the successful treatment of cancers with chemotherapeutic drugs is usually hindered by MDR. Cancer cells with multidrug resistance (MDR) become simultaneously resistant to a variety of structurally and functionally unrelated anticancer drugs.1, 2 Among the various factors contributing to the development of MDR, the overexpression of ABC transporters, particularly ATP-binding cassette subfamily B member 1 (ABCB1), ATP-binding cassette subfamily C member 1 (ABCC1) and ATP-binding cassette subfamily G member 2 (ABCG2), is considered to be a primary determinant of MDR.3, 4 Functionally, ABC transporters can pump chemotherapeutic agent out of cancer cells and decrease the intracellular accumulation of chemotherapeutic agents, resulting in drug resistance in MDR cells.
ABCB1 (P-glycoprotein, P-gp), the first identified eukaryotic ABC transporter that conferred MDR in cancer cells, is composed of two homologous halves, each containing six transmembrane domains and an ATP binding/utilization domain, separated by a flexible polypeptide linker.5 ABCB1 can transport a wide range of hydrophobic compounds, including vinca alkaloids (vinblastine and vincristine), anthracyclines (doxorubicin and daunorubicin), epipodophyllotoxins, taxanes and some tyrosine kinase inhibitors (TKIs), out of cells. Overexpression of ABCB1 is also associated with poor prognosis of cancer patients.6, 7, 8 ABCG2 (breast cancer resistance protein (BCRP), mitoxantrone resistance protein, ATP-binding assette of placenta) is known as a half transporter that effluxes drugs with amphiphilic properties.9, 10 ABCG2 has been reported to mediate MDR to a wide variety of anticancer drugs, including mitoxantrone, topotecan, irinotecan, doxorubicin, nilotinib and imatinib.9, 11, 12, 13 Over the past decades, numerous studies have been devoted to developing ABC transporter inhibitors.14, 15 However, none of these inhibitors has been successful in clinical practice due to poor efficacy and unpredictable adverse effects in patients.16
Fortunately, significant efforts have been made to devise new strategies to overcome MDR to cancer chemotherapy, and the addition of TKIs to combination regimens has gained traction.17 It is known that TKIs can bind to the ATP-binding site of tyrosine kinase, and therefore, it is conceivable that TKIs may bind to the ATP-binding site of ABC transporters. Previous studies have reported that several TKIs, including lapatinib, erlotinib, apatinib and afatinib, were able to modulate the MDR phenotype mediated by ABC transporters and encouraged evaluations of the potential of these TKIs to circumvent the anticancer drug resistance in clinical trials.18, 19, 20, 21, 22, 23
Alectinib, an orally active ATP-competitive anaplastic lymphoma kinase (ALK) inhibitor, was approved by the FDA for the treatment of the patients with ALK-positive NSCLC.1, 21, 24, 25, 26, 27 In clinical trials, alectinib induced promising anti-tumor activity in NSCLC patients with altered ALK activity or crizotinib resistance.28, 29 In this study, we investigated the effect of alectinib on the reversal MDR induced by ABCB1 or ABCG2 in vitro, in vivo and ex vivo.
Materials and methods
Chemicals and reagents
Alectinib was purchased from KareBay Biochem (Monmouth Junction, NJ, USA). The formula of Alectinib is presented in Figure 1a. Antibodies against ABCB1/p-gp (sc-13131), ABCG2 (sc-377176), AKT (sc-8312), p-AKT (sc-7985-R), ERK (sc-514302) and p-ERK (sc-7383) were from Santa Cruz Biotechnology Inc. (Paso Robles, CA, USA). Flow cytometry antibodies against ABCB1 (#557002) and mouse IgG2b/κ (#559532) were purchased from BD Biosciences (San Jose, CA, USA), and ABCG2 (sc-18841) was from Santa Cruz Biotechnology Inc. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was from Kangchen Co. (Shanghai, China). SYBR Green qPCR Master Mix was from ExCell Bio (Shanghai, China). Dulbecco’s modified Eagle’s medium and RPMI 1640 medium were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Rhodamine 123 (Rho 123), 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT), G418, paclitaxel, doxorubicin (DOX), vincristine (VCR), verapamil (VRP), topotecan, mitoxantrone (MX), cisplatin, fumitremorgin C (FTC) and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).

The structure and cytotoxicity of alectinib. The structure of alectinib (a). The expression levels of ABCG2 and ABCB1 were detected in KB cells, ABCB1-overexpressing KBv200 cells, S1 cells and ABCG2-overexpressing S1-MI-80 cells (b). MTT assays were used to assess the cell survival in KB cells and ABCB1-overexpressing KBv200 cells (c), MCF-7 and ABCB1-overexpressing MCF-7/adr cells (d), S1 and ABCG2-overexpressing S1-MI-80 cells (e), H460 and ABCG2-overexpressing H460/MX20 cells (f), HEK293/Vector and stable ABCB1 transfected HEK293/ABCB1 cells (g), and HEK293/Vector and stable ABCG2 transfected HEK293/ABCG2-R2 cells (h). All cells were treated with a full range of alectinib concentrations for 72 h. The data points represent the mean±s.d. of at least three independent experiments performed in quadruple.
Cell lines and cell culture
The human oral epidermoid carcinoma cell line KB and its vincristine (VCR)-selected ABCB1-overexpressing cell line KBv200 were cultured in RPMI 1640 supplemented with 10% FBS at 37 °C in the presence of 5% CO2. The NSCLC cell line H460 and its mitoxantrone (MX)-selected ABCG2-overexpressing cell line H460/MX20, human colon carcinoma cell line S1 and its MX-selected ABCG2-overexpressing cell line S1-MI-80 were generous gifts from Dr Susan Bates (NCI, NIH, Bethesda, MD, USA). The human breast carcinoma cell line MCF-7 and its DOX-selected ABCB1-overexpressing cell line MCF-7/adr; transfected HEK293/Vector, HEK293/ABCB1 and HEK293/ABCG2 (wild type) cells were cultured in Dulbecco’s modified Eagle's medium supplemented with 10% FBS at 37 °C in the presence of 5% CO2. The transfected cells were cultured in medium containing 2 mg ml−1 G418.30, 31, 32, 33
Cell cytotoxicity assay
MTT assays were used to assess cell sensitivity to drugs as previously described.34 The concentration required to inhibit cell growth by 50% (IC50) was calculated from survival curves by the Bliss method.35 The degree of reversal of MDR (fold reversal) was calculated by dividing the IC50 for cells with the chemotherapeutic agent in the absence of alectinib by that obtained in the presence of alectinib.
Animal experiments
The KBv200 cell xenograft model was established as previously described.36 All experiments were performed in accordance with the guidelines for the Use of Laboratory Animals of Sun Yat-sen University Cancer Center after agreement of the Animal Experimental Ethics Review Committee of Sun Yat-sen University Cancer Center. Briefly, KBv200 cells (2.0 × 106) were subcutaneously injected into the right flanks of athymic nude mice (BALB/c-nu/nu, 5–6 weeks of age, 18–24 g in weight). The mice were randomized into four groups and received the following various treatments: (a) saline (q3d); (b) paclitaxel (20 mg kg−1, intraperitoneal injection/i.p., q3d); (c) alectinib (12 mg kg−1, gavage, q3d); (d) alectinib (12 mg kg−1, gavage, q3d, given 1 h before paclitaxel administration) plus paclitaxel (20 mg kg−1, i.p., q3d). When the tumor size reached a mean diameter of 5 mm, the body weights of the animals and the two perpendicular tumor diameters (A and B) were measured every 2 days, and the tumor volume (V) was calculated according to the following formula: V=(π/6)[(A+B)/2]3 × 3. At the end of the observation period, the mice were killed, and the xenografts were excised and weighed. The tumor volume and animal body weight were plotted against the time of treatment. The ratio of tumor growth inhibition (IR) was estimated according to the following formula:37

DOX and Rho 123 accumulation assay
Flow cytometry assays were performed to measure the effect of alectinib on the cellular accumulation of DOX and Rho 123 in the cancer cells as previously described.31 Briefly, the cells were incubated with different concentrations of alectinib or vehicle at 37 °C for 3 h. Following alectinib treatment, 10 μM DOX or 5 μM Rho 123 was added, and incubation was continued for an additional 3 or 0.5 h, respectively. The cells were then harvested, washed three times with chilled phosphate-buffered saline (PBS), and analyzed by flow cytometry (Cytomics FC500, Beckman Coulter Inc., Brea, CA, USA). VRP and fumitremorgin C (FTC) were used as positive control inhibitors of ABCB1 and ABCG2, respectively.
Rho 123 efflux assay
Rho 123 efflux assays were performed as previously described.38 Cells were treated with 5 μM Rho 123 at 37 °C for 30 min. Then, the cells were washed three times with PBS and were then subsequently incubated at 37 °C with fresh media in the presence or absence of 1.5 μM alectinib. The cells were collected at the desired times and immediately washed three times with chilled PBS. Finally, the cells were resuspended in chilled PBS and then immediately subjected to flow cytometric analysis (Cytomics FC500, Beckman Coulte Inc.).
ABCB1 and ABCG2 ATPase activity assays
A colorimetric ATPase assay was performed as previously described with minor modifications.39 Briefly, crude membrane isolated from High Five insect cells expressing either ABCB1 or ABCG2 (100 μg protein per ml) was incubated at 37 °C with a range of different concentrations of alectinib in the presence or absence of sodium orvanadate (0.3 mM for ABCB1 and 1.2 mM for ABCG2) in ATPase assay buffer (50 mM KCl, 5 mM sodium azide, 2 mM EDTA, 10 mM MgCl2, 1 mM DTT, pH 6.8) for 5 min. ATP hydrolysis reaction was then initiated by the addition of 5 mM Mg-ATP (concentration in a final volume of 60 μl) and then incubated for 20 min (for ABCB1) or 10 min (for ABCG2). SDS solution (30 μl of 10% SDS) was added to terminate the reaction. The absorbance was subsequently measured at 750 nm after the addition of a detection reagent (35 mM ammonium molybdate, 15 mM zinc acetate, 10% ascorbic acid) and incubation at 37 °C for 20 min. The amount of inorganic phosphate released was quantified by reading from a standard curve. Specific alectinib-stimulated ABCB1 and ABCG2 ATPase activity (that is, vanadate-sensitive) was determined as the difference between the amounts of inorganic phosphate released from ATP in the absence and presence of sodium orvanadate.
Photo-affinity labeling of ABCB1 and ABCG2 with [125I]-IAAP
Crude membrane from High Five insect cells expressing ABCB1 or ABCG2 (50 μg protein) was incubated with 0–10 μM alectinib for 5 min at room temperature in 50 mM Tris-HCl (pH 7.5). [125I]-IAAP (2200 Ci nmol−1, 3 nM) was added, and incubation was continued for another 5 min under subdued light. The samples were then cross-linked by UV illumination (365 nm) on ice. The labeled ABCB1 and ABCG2 were immunoprecipitated using the corresponding antibody. The samples were then subjected to SDS-polyacrylamide gel electrophoresis using a 7% Tris-acetate NuPAGE gel, dried and exposed to Bio-Max MR film (Eastman Kodak Co., Rochester, NY, USA) at −80 °C overnight. The radioactivity incorporated into the transporter protein was quantified using a Storm 860 Phosphor Imager system (Molecular Dynamics, Sunnyvale, CA, USA).39
Western blot analysis
Western blot analysis was performed as previously described.22 The signal was detected using enhanced chemiluminescence solutions (#1705061, Bio-Rad, CA, USA) and exposed to a Kodak medical X-ray processor (Kodak, Rochester, NY, USA).
PCR and real-time quantitative PCR
ABCB1 and ABCG2 mRNA expression levels were assayed as previously described.22 The PCR primers were 5′-CAGGCTTGCTGTAATTACCCA-3′ (forward) and 5′-TCAAAGAAACAACGGTTCGG-3′(reverse) for ABCB1; 5′-TGGCTGTCATGGCTTCAGTA-3′ (forward) and 5′-GCCACGTGATTCTTCCACAA-3′ (reverse) for ABCG2; 5′-GAGTCAAGGATTTGGTCGT-3′(forward) and 5′-GATCTCGCTCCTGGAAGATG-3′(reverse) for GAPDH. The relative expression of ABCB1 or ABCG2 was quantified using the 2–ΔΔCt method after normalization with GAPDH expression in each sample.40
Cell surface expression of ABCB1 and ABCG2 examined by flow cytometry
Cell surface expression of ABCB1 or ABCG2 in KBv200 or S1-MI-80 cells was measured by flow cytometry after treated with the desired concentrations of alectinib for 48 h. Flow cytometry analysis was performed as previously described.40
Patient samples
Bone marrow samples from diagnosed acute myeloid leukemia (AML) or chronic myelogenous leukemia (CML) patients according to the French–American–British (FAB) classification were obtained after patients provided informed consent.41 This study was approved by the Ethics Review Committee of Sun Yat-sen University. Leukemic blasts were isolated using Ficoll-Hypaque density gradient by centrifugation and then cultured in RPMI 1640 medium.
Statistics
The results are shown as the mean±s.d. All experiments were repeated at least three times, and differences were determined by using Student’s t-test. Statistical significance was considered as *P<0.05, **P<0.01.
Results
Alectinib enhanced the efficacy of conventional substrate chemotherapeutic agents in ABCB1- and ABCG2-overexpressing cells in vitro
It is well known that KBv200 cells and S1-MI-80 cells overexpressing ABCB1 and ABCG2, respectively, (Figure 1b) are extremely resistant to substrate chemotherapeutic agents. To evaluate the reversal effect of alectinib on ABCB1- and ABCG2-mediated MDR in cancer cells, we first tested the cytotoxicity of alectinib alone in different cell lines via MTT assays. The results showed that the IC50 values of alectinib in these cells were all >10 μM and that more than 90% of the cells survived at the concentration of 1.5 μM alectinib (Figure 1c–h). Thus, the concentration of 1.5 μM alectinib was chosen as the maximum concentration in the following studies of reversal MDR.
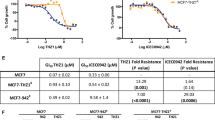
MTT was further performed to confirm the MDR nature of the resistant lines. As expected, the IC50 values of chemotherapeutic agents (such as VCR, DOX, paclitaxel, topotecan) in KBv200 cells, MCF-7/adr cells, S1-MI-80 cells, H460/MX20 cells, HEK293/ABCB1 cells and HEK293/ABCG2 cells were higher than those in their parental sensitive KB cells, MCF-7 cells, S1 cells, H460 cells and HEK293/vector cells (Supplementary Tables 1 and 2). These data suggest that ABCB1- and ABCG2-overexpressing MDR cells were extremely resistant to substrate conventional chemotherapeutic agents compared with their parental sensitive cells. Furthermore, co-treatment with alectinib led to significant decreases in the IC50 values of established ABCB1 and ABCG2 substrates in high-level ABCB1- and ABCG2-expressing cells in, respectively, an alectinib concentration-dependent manner. However, the IC50 values of established ABCB1 and ABCG2 substrates (such as VCR, DOX, paclitaxel and topotecan) in the absence of alectinib showed no significant difference in drug-sensitive cells (Supplementary Tables 1 and 2). In addition, alectinib did not alter the IC50 values of cisplatin, which is not a substrate of ABCB1 or ABCG2 in ABCB1- or ABCG2-overexpressing cells relative to their parental sensitive cells (Supplementary Tables 1 and 2). These results indicate that alectinib can strongly enhance the efficacy of conventional chemotherapeutic agents such as VCR, DOX, paclitaxel and topotecan in ABCB1- and ABCG2-mediated MDR cancer cells in vitro.
Alectinib potentiated the anticancer efficacy of paclitaxel in a model of ABCB1-overexpressing KBv200 cell xenografts in vivo
To investigate whether alectinib could enhance the efficacy of anticancer drugs in vivo, we established a ABCB1-overexpressing multidrug-resistant KBv200 cell xenograft model in nude mice. Paclitaxel has been one of the most commonly used conventional chemotherapeutic agents for oral cancers in the clinic.42 Therefore, we performed KBv200 cell xenografts using paclitaxel. The xenograft growth inhibitions in the alectinib, paclitaxel or combined treatment groups were 1.36%, 11.32% or 48.33%, respectively. There was no significant difference in tumor size among animals treated with saline, alectinib or paclitaxel alone. However, the combination of alectinib and paclitaxel induced a significant greater inhibitory effect on xenograft growth compared with the animals treated with only saline, paclitaxel or alectinib (P<0.01; Figure 2a–c). These data suggest that alectinib can enhance the efficacy of conventional substrate chemotherapeutical agents in ABCB1-overexpressing cell xenografts in vivo. Importantly, at the tested doses, no mortality and a significant decrease in body weight were associated with the combination treatment (Figure 2d), suggesting that the combination regimen did not cause increased toxicity.

Potentiation of the anticancer effects of paclitaxel by alectinib in the KBv200 cell xenograft nude mice model. The tumor growth curve was drawn to monitor the tumor volume with time after implantation. The data shown are expressed as the mean±s.d. of the tumor volume for each group (n=9) (a). The tumor xenografts were excised, and the tumors were photographed on the 21st day after implantation (b). The body weights of the experimental animals were measured every 2 days, and the average percentage change was calculated after treatment (c). The average tumor weight of each group was calculated after the tumors were excised from the mice (d). The data are shown as the mean±s.d. for each group, **P<0.01.
Alectinib increased the intracellular accumulation of DOX and Rho 123 in ABCB1- and ABCG2-overexpressing cells
Alectinib significantly enhanced the sensitivity of MDR cancer cells to conventional substrate chemotherapeutic agents of ABCB1 or ABCG2 in vitro and in vivo. The main cause of ABC transporter-mediated MDR is the decrease in the intracellular accumulation of chemotherapeutic agents by the agents being pumped out of MDR cells. To investigate the potential mechanism, we measured the intracellular accumulation of DOX and Rho 123 in the presence or absence of alectinib in the KBv200 and S1-MI-80 cells by flow cytometric analysis. Our data showed that the intracellular accumulation of DOX and Rho 123 were significantly lower in the resistant cells than that in the parental sensitive cells (Figure 3). However, the intracellular accumulations of DOX and Rho 123 were significantly increased in the presence of alectinib in KBv200 and S1-MI-80 cells but not in the sensitive KB and S1 cells (Figure 3). Moreover, the increased effect produced by alectinib at 1.5 μM was comparable to that by VRP at 10 μM and FTC at 2.5 μM (Figure 3).

Effect of alectinib on the intracellular accumulations of DOX and Rho 123 in MDR cells and in their parental sensitive cells. The intracellular accumulation of DOX (a, b) and Rho 123 (c, d) in KBv200 and S1-MI-80 cells and their parental sensitive KB and S1 cells were measured by flow cytometric analysis, respectively. The results were quantified as the fold change in the fluorescence intensity compared with that obtained in MDR cells. The intensities were calculated by dividing the fluorescence intensity of each sample with that of MDR cells treated with DOX or Rho 123 alone. The data represent the mean±s.d. of at least three independent experiments. *P<0.05, **P<0.01.
To confirm whether the increase in intracellular accumulation of DOX and Rho 123 was due to a blockage of transporter-mediated efflux, the time course of intracellular retention of Rho 123 was examined. As we expected, the intracellular retention of Rho 123 was remarkably less in KBv200 and S1-MI-80 cells than that in their corresponding parental KB and S1 cells in the absence of alectinib (Figure 4a and b). In addition, the intracellular retention of Rho 123 was much more in the presence of alectinib in KBv200 and S1-MI-80 cells than in the absence of alectinib, while there are no significant changes in the presence or absence of alectinib in KB and S1 cells at the same time point (Figure 4a and b). Taken together, the increase in intracellular accumulation of chemotherapeutic agents by alectinib in MDR cells is due to the inhibition of efflux substrate function of ABCB1 and ABCG2.

Effect of alectinib on the efflux of Rho 123, ATPase activity and the [125I]-IAAP photoaffinity labeling of ABCB1 and ABCG2. The cells were pre-incubated with 5 μM Rho 123 at 37 °C for 30 min and then incubated with alectinib for the indicated time points in Rho 123-free media. Rho 123 retention was measured by flow cytometry in KB, KBv200, S1 and S1-MI-80 cells (a, b). The vanadate-sensitive ABCB1 or ABCG2 ATPase activity was evaluated in the presence of the indicated concentrations of alectinib (c, d). Alectinib competed for photolabeling of ABCB1 or ABCG2 by [125I]-IAAP. A representative autoradiogram is shown for three independent experiments. The relative amount of [125I]-IAAP incorporated was plotted against the concentration of alectinib present. Hundred percent incorporation refers to the absence of alectinib (e, f). The data points represent the mean±s.d. of at least three independent experiments. **P<0.01.
Alectinib interacted with the drug-substrate-binding sites of ABCB1 and ABCG2 to stimulate their ATPase activity
As ABC transporters use energy generated from ATP hydrolysis to pump their substrate agents out of MDR cells against a concentration gradient, ATP consumption thus reflects ATPase activity. To explore the mechanism of the inhibition of ABCB1 and ABCG2 function by alectinib, we first evaluated vanadate-sensitive ABCB1 or ABCG2 ATPase activity in the presence of a range of concentrations of alectinib. ABCB1 ATPase activity was stimulated by alectinib until it reached a plateau near 55 nmol min−1 per mg protein, and it remained steady at alectinib concentrations >5 μM. However, a maximum ABCG2 ATPase activity up to 70 nmol min−1 per mg protein was attained in the presence of 1.25 μM alectinib. At higher concentrations of alectinib, a drop in the stimulated ABCG2 ATPase activity was observed (Figure 4c and d). It was observed that alectinib stimulated the ATPase activity similar to other substrates by interacting with the drug-substrate-binding sites on the transporters.
It has been reported that the substrates or inhibitors of ABCB1 or ABCG2 can compete with [125I]-IAAP for photolabeling of the respective transporters.41 Therefore, to further investigate the interaction of alectinib with the substrate-binding sites of ABCB1 and ABCG2, crude membranes from High Five insect cells expressing ABCB1 or ABCG2 were incubated with [125I]-IAAP and alectinib (0–10 μM). The autoradiogram showed the inhibition of photolabeling of ABCB1 or ABCG2 with [125I]-IAAP by alectinib in a concentration-dependent manner (Figure 4e and f). The concentrations required for 50% inhibition (IC50) of photolabeling of ABCB1 or ABCG2 with [125I]-IAAP were 2.5 or 0.6 μM, respectively. These data demonstrated that alectinib, similar to IAAP, interacts at the drug-binding pocket of ABCB1 and ABCG2. Thus, alectinib might compete with substrates to interact with the substrate-binding sites of ABCB1 and ABCG2, leading to inhibitory efflux activity of the transporters.
Alectinib alters neither the expression levels nor the cell surface localization of ABCB1 and ABCG2
The reversal of ABCB1- and ABCG2-mediated drug resistance by alectinib could be achieved by either antagonizing the function or decreasing the expression levels of the transporters. Therefore, the effect of alectinib on the protein and mRNA expression levels of ABCB1 and ABCG2 was detected by western blot and RT-PCR assays, respectively. Alectinib did not alter the protein level of either transporter, even at concentrations up to 15.0 μM (Figure 5a and b). The mRNA expression of ABCB1 or ABCG2 was also unchanged (Figure 5c and d). In addition, we further performed a flow cytometric assay to determine the effect of alectinib on the cell surface localization of ABCB1 and ABCG2. Alectinib influenced neither the cell surface expression nor the intracellular localization of ABCB1 and ABCG2 (Figure 5e and f). Thus, these results suggest that the enhancement of chemotherapeutic agents by alectinib is neither associated with the expression levels of protein and mRNA nor the localization of ABCB1 or ABCG2 protein.

Effect of alectinib on the expression levels of ABCB1 or ABCG2 in MDR cells. The protein levels of ABCB1 and ABCG2 were detected by western blot (a, b). Real-time PCR assay was performed to quantify the mRNA levels of ABCB1 and ABCG2 in the MDR cells (c, d). The cell surface expression of ABCB1 or ABCG2 in KBv200 or S1-MI-80 cells, respectively, was detected by flow cytometry (e, f). Representative results from three independent experiments are shown in a, b, e and f, respectively.
Alectinib did not block the phosphorylation of AKT and ERK in the concentration of reversal MDR
Some studies have reported that blocking the phosphorylation of AKT or ERK could increase cancer cell sensitivity to chemotherapeutic agents.43, 44 Alectinib is a potent ALK and c-Met inhibitor that regulates several important downstream signaling molecules such as AKT and ERK. As we did not detect the expression of ALK in KBv200 and S1-MI-80 cells and their parental sensitive cells, we investigated whether alectinib altered the phosphorylation level of c-Met and its downstream molecules AKT or ERK in the concentration of reversal MDR by western blots. Alectinib altered neither the phosphorylation nor total levels of c-Met, AKT and ERK in KBv200 and S1-MI-80 cells, and their parental sensitive cells in the concentration of reversal MDR (Figure 6). These suggested that the reversal of MDR by alectinib did not involve the blockade of phosphorylation of c-Met, AKT and ERK.

Effect of alectinib on the inhibition of AKT, ERK and c-Met phosphorylation. The protein levels of AKT, ERK and c-Met were detected by western blot in KB (a, e), KBv200 (b, f), S1 (c, g) and S1-MI-80 (d, h) cells treated with alectinib at the indicated concentrations and designated time points. Alectinib (15.0 μM) was used as a positive control for the blockage of AKT, ERK and c-Met phosphorylation. Representative results from three independent experiments are shown in each panel.
Alectinib effectively reversed MDR in ex vivo
It has been reported that ABCB1 is expressed in both AML and CML.45, 46 To investigate whether alectinib could reverse ABCB1-mediated MDR in ex vivo, bone marrow samples were collected from resistant patients with AML or CML. Four of the eleven patient samples exhibited high expression of ABCB1 (Figure 7a). We then examined the effect of alectinib on the intracellular accumulation of Rho 123 in these ABCB1-overexpressing primary leukemia blast samples using flow cytometric analysis. As shown in Figure 7b and c, alectinib significantly increased the intracellular accumulation of Rho 123. In addition, we used MTT assays to assess the sensitization effect of alectinib in these samples. The result revealed that 1.5 μM alectinib significantly increased the sensitivity of all four samples to DOX, which exhibited similar efficiency to 10 μM VRP (Figure 7d). These results suggest that alectinib is able to sensitize ABCB1-overexpressing primary leukemia cells to conventional anticancer drugs, and thus, it might be useful in combination regimens to combat ABCB1-mediated MDR in the clinic. The schematic model illustrating the reversal of MDR by alectinib was showed in Figure 8.

Alectinib increased the accumulation of Rho 123 and enhanced the cytotoxicity of DOX in ABCB1-overexpressing primary leukemia blasts. Bone marrow was collected from four newly diagnosed AML/CML patients. Mononuclear cells were isolated, and their expression levels of ABCB1 were assessed by flow cytometry (a). Intracellular accumulation of Rho 123 in primary leukemia blasts with or without alectinib treatment was analyzed by flow cytometry. VRP, an ABCB1 inhibitor, was used as a positive control (b). The results were calculated by dividing the fluorescence intensity of each sample with that of mononuclear cells treated with Rho 123 alone (c). Enhancement of DOX cytotoxicity in ABCB1-overexpressing primary leukemia blasts by alectinib (d). The data represent the mean±s.d. of at least three independent experiments. Representative results are shown in a and b. *P<0.05, **P<0.01.

A schematic model illustrating the reversal of MDR by alectinib. When the primary tumors were treated with chemotherapeutic agents, the sensitive cancer cells were killed, but the MDR cancer cells simultaneously became resistant to structurally and mechanistically distinct classes of anticancer drugs. Overexpressing ABCB1 or ABCG2 in the MDR cells resulted in an efflux of drugs across the membrane. In the presence of alectinib, the intracellular accumulation of conventional anticancer drugs was increased in the ABCB1- or ABCG2-overexpressing cells, resulting in sensitization of the MDR cells. The reversal of MDR by alectinib was mediated by inhibition of ABCB1/ABCG2 transport function and by enhanced intracellular accumulation of anticancer substrate drugs.
Discussion
Although chemotherapy offers clinical benefits in many cancers, its impact on chemo-resistant patients is still limited, likely due to MDR, which prevents a full beneficial response. The two most variously studied MDR transporters, ABCB1 and ABCG2, are known to be highly expressed in cancer cells with MDR phenotypes and actively efflux drugs. It is therefore logical to speculate that developing and identifying modulators that regulate these transporters or block their function may provide an effective strategy to circumvent MDR.
Currently, two approved ALK inhibitors, crizotinib and ceritinib, have been reported to have potent reversing activity in ABCB1- or ABCG2-overexpressing MDR cells in vitro and in vivo,47, 48 suggesting that ALK inhibitors may have an important interaction with ABC transporters, such as ABCB1 and ABCG2. Here we showed for the first time that alectinib significantly potentiated the efficacy of substrate chemotherapeutic agents in ABCB1- or ABCG2-overexpressing cells under the more than 90% cell survival concentration of alectinib, while it did have any effect on the chemotherapeutic sensitivity of their parental cells. More importantly, alectinib did not enhance the cytotoxicity of cisplatin (non-substrate of ABCB1 and ABCG2) in ABCB1- or ABCG2-overexpressing cells. Alectinib also remarkably potentiated the anticancer efficacy of paclitaxel in the ABCB1-overexpressing KBv200 cell xenograft model. Thus, alectinib specifically enhanced the efficacy of chemotherapeutic agents in ABCB1- or ABCG2-overexpressing cells in vitro and in vivo.
Consistent with the enhancement of chemotherapeutic agents by alectinib in vitro and in vivo, alectinib was found to increase the intracellular accumulation of DOX and Rho 123 in ABCB1- or ABCG2-overexpressing cells. This effect was achieved by inhibiting the efflux function of the ABCB1 or ABCG2 transporter. The stimulation of ATPase activity of ABCB1 and ABCG2 by alectinib at low concentrations suggests that alectinib may be similar to their substrates. In other words, alectinib seems to be a competitive inhibitor of these transporters. The interactions of alectinib to ABCB1 and ABCG2 were further confirmed by its competition with the [125I]-IAAP photoaffinity labeling of the transporters. These results are consistent with our earlier findings with other TKIs.18, 20
It was reported that the blockage of AKT or ERK phosphorylation could increase cancer cell sensitivity to anticancer drugs such as cisplatin.43, 44 Some TKIs such as gefitinib and afatinib can also downregulate ABCG2 expression.18, 37 Because alectinib is an ALK and c-Met TKI, it could inhibit phosphorylation of signals downstream AKT and ERK in ALK- or c-Met-positive NSCLC cells.26 However, alectinib altered neither the mRNA and protein expression levels and localization of ABCB1/ABCG2 nor the phosphorylation levels of c-Met, AKT and ERK at the reversal MDR concentrations. These suggest that the reversal of MDR by alectinib results from the inhibition of ABCB1/ABCG2 transport function but does not involve in the alteration of their expression levels of mRNA and protein or membrane localization.
Treatment failure in AML is frequently due to resistance to chemotherapy through different mechanisms, in which drug efflux via ATP-binding cassette (ABC) proteins, specifically ABCB1, is one of the most recognized. It has been reported that 23 of 63 (28.6%) of newly treated patients and 11 of 22 (59.1%) of relapsed/refractory patients exhibited high ABCB1 expression. In primary leukemia cells ex vivo, 4 of 11 (36.3%) of leukemic blast samples overexpressed ABCB1. Importantly, alectinib significantly increased the accumulation of Rho 123 and enhanced the cytotoxicity of DOX in these ABCB1-overexpressing primary leukemic blast cells. Consequently, these results suggest that alectinib can reverse ABCB1-mediated MDR ex vivo, which encourages combinational therapies of alectinib and conventional chemotherapeutic drugs in cancer patients with ABCB1-mediated MDR and deserves further study in the clinic.
In conclusion, we provide the first in vitro, in vivo and ex vivo evidence that alectinib significantly enhances the efficacy of chemotherapeutic agents in ABCB1- and ABCG2-overexpressing cells. The reversal of MDR by alectinib was mediated by inhibition of ABCB1/ABCG2 transport function, resulting in an increase in the intracellular accumulation of substrate chemotherapeutic drugs. These positive finding suggest that combinational therapies of alectinib with conventional ABCB1 or ABCG2 substrate chemotherapeutic drugs may benefit patients with ABCB1- or ABCG2-mediated MDR.
References
Diamond EL, Durham BH, Haroche JL, Yao Z, Ma J, Parikh SA et al. Alectinib approved for ALK+ lung cancer. Cancer Discov 2016; 6: 115.
Gottesman MM . Mechanisms of cancer drug resistance. Annu Rev Med 2002; 53: 615–627.
Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD . Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat 2003; 6: 71–84.
Dean M, Hamon Y, Chimini G . The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 2001; 42: 1007–1017.
Fukuda Y, Schuetz JD . ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem Pharmacol 2012; 83: 1073–1083.
Lage H . MDR1/P-glycoprotein (ABCB1) as target for RNA interference-mediated reversal of multidrug resistance. Curr Drug Targets 2006; 7: 813–821.
Jamroziak K, Robak T . Pharmacogenomics of MDR1/ABCB1 gene: the influence on risk and clinical outcome of haematological malignancies. Hematology 2004; 9: 91–105.
Leonard GD, Fojo T, Bates SE . The role of ABC transporters in clinical practice. Oncologist 2003; 8: 411–424.
Mao Q, Unadkat JD . Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—an update. AAPS J 2015; 17: 65–82.
Kathawala RJ, Chen JJ, Zhang YK, Wang YJ, Patel A, Wang DS et al. Masitinib antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Int J Oncol 2014; 44: 1634–1642.
Shukla S, Kouanda A, Silverton L, Talele TT, Ambudkar SV . Pharmacophore modeling of nilotinib as an inhibitor of ATP-binding cassette drug transporters and BCR-ABL kinase using a three-dimensional quantitative structure-activity relationship approach. Mol Pharm 2014; 11: 2313–2322.
Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR Jr, Chen X et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol 2009; 78: 153–161.
Wang L, Giannoudis A, Lane S, Williamson P, Pirmohamed M, Clark RE . Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin Pharmacol Ther 2008; 83: 258–264.
Kathawala RJ, Wang YJ, Ashby CR Jr, Chen ZS . Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin J Cancer 2014; 33: 223–230.
Deng W, Dai CL, Chen JJ, Kathawala RJ, Sun YL, Chen HF et al. Tandutinib (MLN518) reverses multidrug resistance by inhibiting the efflux activity of the multidrug resistance protein 7 (ABCC10). Oncol Rep 2013; 29: 2479–2485.
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM . Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5: 219–234.
Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett 2013; 328: 307–317.
Wang XK, To KK, Huang LY, Xu JH, Yang K, Wang F et al. Afatinib circumvents multidrug resistance via dually inhibiting ATP binding cassette subfamily G member 2 in vitro and in vivo. Oncotarget 2014; 5: 11971–11985.
Ma SL, Hu YP, Wang F, Huang ZC, Chen YF, Wang XK et al. Lapatinib antagonizes multidrug resistance-associated protein 1-mediated multidrug resistance by inhibiting its transport function. Mol Med 2014; 20: 390–399.
Mi YJ, Liang YJ, Huang HB, Zhao HY, Wu CP, Wang F et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res 2010; 70: 7981–7991.
Shi Z, Parmar S, Peng XX, Shen T, Robey RW, Bates SE et al. The epidermal growth factor tyrosine kinase inhibitor AG1478 and erlotinib reverse ABCG2-mediated drug resistance. Oncol Rep 2009; 21: 483–489.
Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res 2008; 68: 7905–7914.
Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res 2007; 67: 11012–11020.
Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C et al. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol 2014; 9: 549–553.
Latif M, Saeed A, Kim SH . Journey of the ALK-inhibitor CH5424802 to phase II clinical trial. Arch Pharm Res 2013; 36: 1051–1054.
Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011; 19: 679–690.
Song Z, Wang M, Zhang A . Alectinib: a novel second generation anaplastic lymphoma kinase (ALK) inhibitor for overcoming clinically-acquired resistance. Acta Pharm Sin B 2015; 5: 34–37.
Klempner SJ, Ou SH . Anaplastic lymphoma kinase inhibitors in brain metastases from ALK+ non-small cell lung cancer: hitting the target even in the CNS. Chin Clin Oncol 2015; 4: 20.
Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol 2014; 15: 1119–1128.
Zhang JY, Wu HY, Xia XK, Liang YJ, Yan YY, She ZG et al. Anthracenedione derivative 1403P-3 induces apoptosis in KB and KBv200 cells via reactive oxygen species-independent mitochondrial pathway and death receptor pathway. Cancer Biol Ther 2007; 6: 1413–1421.
Fu L, Liang Y, Deng L, Ding Y, Chen L, Ye Y et al. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother Pharmacol 2004; 53: 349–356.
Robey RW, Shukla S, Finley EM, Oldham RK, Barnett D, Ambudkar SV et al. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R)). Biochem Pharmacol 2008; 75: 1302–1312.
Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer 2003; 89: 1971–1978.
Chen LM, Wu XP, Ruan JW, Liang YJ, Ding Y, Shi Z et al. Screening novel, potent multidrug-resistant modulators from imidazole derivatives. Oncol Res 2004; 14: 355–362.
Shi Z, Liang YJ, Chen ZS, Wang XW, Wang XH, Ding Y et al. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol Ther 2006; 5: 39–47.
Chen LM, Liang YJ, Ruan JW, Ding Y, Wang XW, Shi Z et al. Reversal of P-gp mediated multidrug resistance in-vitro and in-vivo by FG020318. J Pharm Pharmacol 2004; 56: 1061–1066.
Pick A, Wiese M . Tyrosine kinase inhibitors influence ABCG2 expression in EGFR-positive MDCK BCRP cells via the PI3K/Akt signaling pathway. ChemMedChem 2012; 7: 650–662.
Dai CL, Liang YJ, Wang YS, Tiwari AK, Yan YY, Wang F et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett 2009; 279: 74–83.
Ambudkar SV . Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol 1998; 292: 504–514.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Tong XZ, Wang F, Liang S, Zhang X, He JH, Chen XG et al. Apatinib (YN968D1) enhances the efficacy of conventional chemotherapeutical drugs in side population cells and ABCB1-overexpressing leukemia cells. Biochem Pharmacol 2012; 83: 586–597.
Mody MD, Gill HS, Saba NF . The evolving and future role of taxanes in squamous cell carcinomas of the head and neck: a review. JAMA Otolaryngol Head Neck Surg 2016; 142: 898–905.
Dey A, Wong E, Kua N, Teo HL, Tergaonkar V, Lane D . Hexamethylene bisacetamide (HMBA) simultaneously targets AKT and MAPK pathway and represses NF kappaB activity: implications for cancer therapy. Cell Cycle 2008; 7: 3759–3767.
Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, Billi AM et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia 2008; 22: 147–160.
Pera B, Calvo-Vidal MN, Ambati S, Jordi M, Kahn A, Diaz JF et al. High affinity and covalent-binding microtubule stabilizing agents show activity in chemotherapy-resistant acute myeloid leukemia cells. Cancer Lett 2015; 368: 97–104.
Gao F, Dong W, Yang W, Liu J, Zheng Z, Sun K . Expression of P-gp in acute myeloid leukemia and the reversal function of AsO on drug resistance. Oncol Lett 2015; 9: 177–182.
Hu J, Zhang X, Wang F, Wang X, Yang K, Xu M et al. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Oncotarget 2015; 6: 44643–44659.
Zhou WJ, Zhang X, Cheng C, Wang F, Wang XK, Liang YJ et al. Crizotinib (PF-02341066) reverses multidrug resistance in cancer cells by inhibiting the function of P-glycoprotein. Br J Pharmacol 2012; 166: 1669–1683.
Acknowledgements
We thank Dr Susan Bates (NCI, NIH, Bethesda, MD) for the ABCG2-overexpressing cell lines. This work was supported by grants from the National High Technology Research and Development Program of China (863 project, no. 2012AA02A303) and the International Collaboration Science Research Foundation of Guangdong Province (no. 2013B051000046).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yang, K., Chen, Y., To, K. et al. Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. Exp Mol Med 49, e303 (2017). https://doi.org/10.1038/emm.2016.168
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2016.168
This article is cited by
-
Adagrasib, a KRAS G12C inhibitor, reverses the multidrug resistance mediated by ABCB1 in vitro and in vivo
Cell Communication and Signaling (2022)
-
Mechanistic basis of breast cancer resistance protein inhibition by new indeno[1,2-b]indoles
Scientific Reports (2021)
-
Apatinib Mesylate in the treatment of advanced progressed lung adenocarcinoma patients with EGFR-TKI resistance —A Multicenter Randomized Trial
Scientific Reports (2019)
-
Clinical Pharmacokinetics of Anaplastic Lymphoma Kinase Inhibitors in Non-Small-Cell Lung Cancer
Clinical Pharmacokinetics (2019)
-
Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells
Molecular Cancer (2018)
