
Abstract
A limited number of microRNAs (miRNAs, miRs) have been reported to control postnatal cardiomyocyte proliferation, but their strong regulatory effects suggest a possible therapeutic approach to stimulate regenerative capacity in the diseased myocardium. This study aimed to investigate the miRNAs responsible for postnatal cardiomyocyte proliferation and their downstream targets. Here, we compared miRNA profiles in cardiomyocytes between postnatal day 0 (P0) and day 10 (P10) using miRNA arrays, and found that 21 miRNAs were upregulated at P10, whereas 11 were downregulated. Among them, miR-31a-5p was identified as being able to promote cardiomyocyte proliferation as determined by proliferating cell nuclear antigen (PCNA) expression, double immunofluorescent labeling for α-actinin and 5-ethynyl-2-deoxyuridine (EdU) or Ki-67, and cell number counting, whereas miR-31a-5p inhibition could reduce their levels. RhoBTB1 was identified as a target gene of miR-31a-5p, mediating the regulatory effect of miR-31a-5p in cardiomyocyte proliferation. Importantly, neonatal rats injected with a miR-31a-5p antagomir at day 0 for three consecutive days exhibited reduced expression of markers of cardiomyocyte proliferation including PCNA expression and double immunofluorescent labeling for α-actinin and EdU, Ki-67 or phospho-histone-H3. In conclusion, miR-31a-5p controls postnatal cardiomyocyte proliferation by targeting RhoBTB1, and increasing miR-31a-5p level might be a novel therapeutic strategy for enhancing cardiac reparative processes.
Similar content being viewed by others


Introduction
Adult mammalian cardiomyocytes retain some limited endogenous renewal capacity; however, this capacity is insufficient for the replacement of acute or chronic cardiomyocyte loss in ischemic cardiac injury or heart failure.1 Unlike the adult heart, the neonatal mammalian heart is able to substantially regenerate after injury by increasing the level of cardiomyocyte proliferation, although this regenerative capacity is lost by postnatal day 7.2 In addition, cardiomyocyte proliferation has been reported to contribute to developmental heart growth in young humans.3 Interestingly, adult mammalian cardiomyocytes withdraw from the cell cycle soon after birth (3–5 days after birth in rats).4, 5 A better understanding of the underlying mechanisms for the transition of cardiomyocytes from a proliferative phenotype to a quiescent state would help identify potential novel strategies to reactivate the dormant regenerative capacity in the adult heart upon injury.4, 6
MicroRNAs (miRNAs, miRs) are small, noncoding RNAs that have fundamental roles in almost all aspects of the gene regulatory network.7, 8 miRNAs are capable of regulating the cell phenotype, including proliferation and hypertrophy,7, 9 and are necessary for embryonic and neonatal heart development, as well as cardiac homeostasis in adults.10, 11 Dysregulation of miRNAs has been reported in myocardial infarction, fibrosis, hypertrophy and heart failure.10, 12, 13, 14, 15
Accumulating evidence suggests that dysregulated miRNAs such as miR-195, -29a, -30a and -141 contribute to the postnatal mitotic arrest of cardiomyocytes.5, 16, 17 These studies either used whole ventricles or investigated a later time point (4 weeks or 1 year after birth) in purified cardiomyocytes.5, 16, 17 As cardiomyocyte proliferation drops quickly after birth and after primary isolation,6, 16 we compared the miRNA profiles in cardiomyocytes between postnatal day 0 (P0) and day 10 (P10), an earlier time point than previously examined.
Here, we found 32 miRNAs that were differentially expressed in cardiomyocytes between postnatal day 0 (P0) and day 10 (P10) based on miRNA arrays. Although miR-31a-5p was significantly upregulated in P10 cardiomyocytes compared with its expression in P0 cardiomyocytes, it was surprisingly found to promote cardiomyocyte proliferation, whereas inhibition of miR-31a-5p decreased proliferation, which is contrary to our initial hypothesis. We consider the upregulation of miR-31a-5p in P10 cardiomyocytes as a compensatory mechanism of the cardiomyocyte in response to exiting the cell cycle. RhoBTB1 was identified as a target gene of miR-31a-5p responsible for the pro-proliferative effect of miR-31a-5p in cardiomyocytes. Finally, neonatal rats injected with miR-31a-5p antagomir at day 0 for 3 days exhibited reduced expression of cardiomyocyte proliferation markers, indicating that miR-31a-5p is required for postnatal cardiomyocyte proliferation in vivo.
Materials and methods
Neonatal rat ventricular cardiomyocyte isolation
All rats were purchased and raised in the Experimental Animal Center of Nanjing Medical University (Nanjing, China). All procedures with rats were in accordance with guidelines on the use and care of laboratory animals for biomedical research published by the National Institutes of Health (No. 85-23, revised 1996), and the experimental protocol was reviewed and approved by the Animal Care and Use Committee of Nanjing Medical University. Primary neonatal rat ventricular cardiomyocytes (NRVMs) using ventricles from day 0 (P0) and day 10 (P10) of neonatal pups were prepared as previously described18 and purified by Percoll gradient centrifugation.
RNA isolation and miRNA arrays
Total RNA (including miRNA) was extracted from cardiomyocytes using the RNeasy Mini Kit (Qiagen, Hilden, Germany). miRNA profiling was performed using OE Biotech’s (Shanghai, China) miRNA microarray service. The MIAME-compliant data have been submitted to the Gene Expression Omnibus (GEO, platform ID: GSE74205).
Quantitative reverse transcription polymerase chain reaction
To validate the miRNA arrays results, the Bulge-Loop miRNA qPCR Primer Set (RiboBio, Guangzhou, China) was used to determine the expression levels of dysregulated miRNAs by Quantitative reverse transcription polymerase chain reactions (qRT-PCRs) with iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) on an ABI-7900 Real-Time PCR Detection System (7900HT, Applied Biosystems, Foster city, CA, USA). U6 was used as an endogenous control. The expression levels of RhoBTB1, RhoBTB2 and RhoBTB3 were analyzed by quantitative PCRs with SYBR Green (TaKaRa) on an ABI-7900 Real-Time PCR Detection System (7900HT, Applied Biosystems). 18S was used as an inner control for normalization. The primer sequences used were as follows (5′–3′): RhoBTB1 (forward GAAAGCCTTCCACGTCAGGA and reverse GTGCAACCAAGTGTGTCAGG), RhoBTB2 (forward TCTTCGAGCCTCCATGACATT and reverse ACCTCCTAGGGTCCCAGTTC) and RhoBTB3 (forward GTAGCTTGTTGCTGAACGCC and reverse GCCGCAATGATGACTGGAAC). The 18S (forward AGTCCCTGCCCTTTGTACACA and reverse CGATCCGAGGGCCTCACTA) was used for normalization. The relative expression level was calculated using the 2−ΔΔCt method.
Cell transfection and cell proliferation assay
MicrON miRNA mimic (50 nM), micrOFF miRNA inhibitor (100 nM) or their negative controls (RiboBio, China) were transfected into cardiomyocytes for 48 h using Lipofectamine 2000 (Invitrogen, Cambridge, MA, USA), as recommended by the manufacturer. Transfection with siRNAs for RhoBTB1 (75 μM) (Invitrogen) was carried out using Lipofectamine 2000. For 5-ethynyl-2-deoxyuridine (EdU) staining, 24 h after transfection with the miRNA mimic (incubated with 1% serum), inhibitor (incubated with 10% serum) or their negative controls, NRVMs were labeled with EdU for 24 h before harvesting, and the Click-iT EdU Alexa Fluor 555 Imaging Kit (Thermo Scientific, Cambridge, MA, USA) was used to reveal EdU incorporation. For Ki-67 staining, NRVMs were incubated with the following primary antibodies overnight: α-actinin (1:100, A7811, Sigma-Aldrich, St Louis, MO, USA) and Ki-67 (1:100, ab 16667, Abcam, Cambridge, UK).
Luciferase reporter assay
Complementarity between RhoBTB1-3′UTR and the rno-miR-31a-5p sequence was obtained from RNAhybrid (V2.1) (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/). A pmiR-RB-REPORT vector (Ribobio) was constructed by inserting a fragment of the 3′-UTR of RhoBTB1 mRNA containing the putative miR-31a-5p binding site as follows: r_Rhobtb1_3UTR_F: GCGCTCGAGCCGTTGGATAACC GTGTC and r_Rhobtb1_3UTR_R: AATGCGGCCGCTTTGGCAAGAATACAATC, using the XhoI and NotI sites. As a mutated control vector, a 3′-UTR fragment, with mutations in the seed binding sites, was generated using the following primer: r_Rhobtb1_mut_R1937 (NotI): AATGCGGCCGCTTTCCGTTCTATACAATC. For reporter assays, 293T cells were transfected with the wild-type or mutant reporter plasmid and rno-miR-31a-5p or negative control mimics (RiboBio). Firefly and Renilla luciferase activities were measured 48 h post-transfection using the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Western blotting
Equal amounts of protein were subjected to SDS-PAGE and transferred onto PVDF membranes. Antibodies against RhoBTB1 (1:1000; ab88876, Abcam, Cambridge, UK) and PCNA (1:1000; 13110, Cell Signaling Technology, Boston, MA, USA) were used as primary antibodies. Mouse or rabbit IgG antibodies coupled to horseradish peroxidase (HRP) were used as secondary antibodies. Actin (1:1000; cat. 4967, Cell Signaling Technology) was used as the loading control. The blots were developed with an enhanced chemiluminescence reagent (Thermo Scientific) and exposed on a ChemiDoc MP imager (Bio-Rad Laboratories). Image Lab software was used to quantify the band density.
miR-31a-5p antagomir injections and immunofluorescence staining
miR-31a-5p antagomir (2‘OME+ 5’chol modified) (RiboBio) injections were performed. To inhibit miR-31a-5p in neonatal rats, 50 mg kg−1 antagomir or the scramble control was given intraperitoneally for 3 consecutive days after birth, and 5-ethynyl-2′-deoxyuridine (EdU, 50 mg kg−1) was injected intraperitoneally daily 2 days before harvesting at P4. Ventricular tissues were snap frozen in liquid nitrogen with OCT on the short axis at 8 μm. For staining, sections were fixed in 4% paraformaldehyde (PFA) followed by washing with PBS. Sections were blocked with 3% (w/v) BSA in PBS and then incubated with primary antibodies overnight: p-histone H3 (1:100, ab5176, Abcam), Ki-67 (1:100, ab 16667, Abcam) and α-actinin (1:100, A7811, Sigma-Aldrich).
Statistical analysis
Data were presented as the mean±s.e. An unpaired t-test was used for comparisons between two groups with a significance level of 0.05. A one-way ANOVA test was performed to compare values among more than three groups followed by Bonferroni’s tests to compare the differences between every two groups. P-values <0.05 were considered to be statistically significant. All analyses were performed using GraphPad Prism 5.
Results
miR-31a-5p controls postnatal NRVM proliferation in vitro
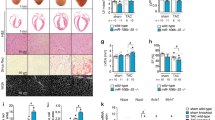
To identify miRNAs responsible for postnatal NRVM proliferation, we compared the miRNA expression in cardiomyocytes isolated from rat pups at 0 day and 10 days of age. A total of 32 miRNAs were differentially expressed between cardiomyocytes isolated from these two time points, among which 21 miRNAs were upregulated and 11 were downregulated at P10 (Figure 1a and Table 1). As miR-31a-3p was the most upregulated miRNA from these array results and miR-31a-5p was localized in its 5p-arm (Table 1), we selected these two miRNAs for further analysis of their functional role in NRVM proliferation. First, we validated our miRNA array data by qRT-PCR and demonstrated the upregulation of both of these selected miRNAs in cardiomyocytes at 10 days compared with their expression at P0 (Figure 1b). Next, we assessed the effects of the gain-of-function of these miRNAs on EdU incorporation (a measure of cellular proliferation) in NRVMs. NRVMs (isolated from 0-day rat pups) transfected with miR-31a-3p did not exhibit an increase in EdU incorporation (Figure 1c), whereas miR-31a-5p transfection increased the expression of EdU-positive cardiomyocytes (Figure 1d). The effect of miR-31a-5p on NRVM proliferation was contrary to our initial hypothesis, and we considered the upregulation of miR-31a-5p in P10 cardiomyocytes as a compensatory response. NRVM proliferation as assayed by EdU incorporation was also confirmed by Ki-67 immunostaining, another marker for cellular proliferation (Figure 1e), cell counting to measure the total number of cardiomyocytes (Figure 1f), and the expression level of PCNA as shown by western blotting (Figure 1g). Taken together, these data suggest that miR-31-5p is sufficient to induce NRVM proliferation.

miR-31a-5p promotes cardiomyocyte proliferation in vitro. (a) MicroRNA (miRNA) arrays identified 32 differentially expressed miRNAs between cardiomyocytes isolated from rats at 0 day and 10 days of age. n=4 per group. (b) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of miR-31a-3p and miR-31a-5p between cardiomyocytes isolated from rats at 0 day and 10 days of age. n=4 per group. (c) Immunohistochemical stainings for sarcomeric α-actinin and 5-ethynyl-2-deoxyuridine (EdU) staining were determined when neonatal rat ventricular cardiomyocytes (NRVMs) were transfected with the control (NC mimic) or the miR-31a-3p mimic. At least 2000 cells were quantified in each group. (d–f) Immunohistochemical stainings for sarcomeric α-actinin and EdU (d) or Ki-67 (e) followed by quantification of the cell number (f) were performed when NRVMs were transfected with the control (NC mimic) or the miR-31a-5p mimic. At least 2000 cells were quantified in each group. Scale bar: 100 μm. (g) Western blot showed that proliferating cell nuclear antigen (PCNA) expression was increased by the miR-31a-5p mimic. n=3 per group. *P<0.05, **P<0.01 versus the respective control.
Next, we tested the effect of miR-31a-5p loss-of-function on NRVM proliferation. Transfection of 0-day NRVMs with the miR-31-5p inhibitor led to a marked decrease in miR-31-5p levels (Figure 2a). Inhibition of miR-31-5p in these cardiomyocytes led to a decrease in NRVM proliferation as assayed by EdU incorporation (Figure 2b), Ki-67-positive cells (Figure 2c) and total number of NRVMs by cell counting (Figure 2d). Finally, the PCNA level as assessed by western blotting was also decreased by inhibition of miR-31-5p in NRVMs (Figure 2e). These experiments suggested that miR-31a-5p was necessary for NRVM proliferation in 0-day postnatal NRVMs.

Inhibition of miR-31a-5p attenuates cardiomyocyte proliferation in vitro. (a) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of miR-31a-5p in cells transfected with its inhibitor. n=4 per group. (b–d) Immunohistochemical stainings for sarcomeric α-actinin and 5-ethynyl-2-deoxyuridine (EdU) (b) or Ki-67 (c) followed by quantification of the cell number (d) were performed when neonatal rat ventricular cardiomyocytes (NRVMs) were transfected with the control (NC inhibitor) or miR-31a-5p inhibitor. At least 2000 cells were quantified in each group. Scale bar: 100 μm. (e) Western blot showed that PCNA expression was decreased by the miR-31a-5p inhibitor. *P<0.05, **P<0.01, ***P<0.001 versus the respective control.
RhoBTB1 is a target gene of miR-31a-5p
Bioinformatics analysis using Targetscan indicated RhoBTB1 as a potential target gene of miR-31a-5p. Using a reporter construct with the putative 3′-UTR miR-31a-5p binding site of RhoBTB1 downstream of the luciferase gene, we showed that miR-31a-5p led to a reduction in luciferase activity, providing experimental validation of RhoBTB1 as a target for miR-31-5p. Accordingly, after mutation of the binding site, the luciferase activity could not be altered, confirming that the binding of miR-31-5p to its 3′-UTR site was necessary for the silencing of RhoBTB1 (Figure 3a). Western blotting confirmed that the miR-31a-5p mimic downregulated, whereas its inhibitor upregulated RhoBTB1 expression in cultured NRVMs (Figure 3b and c), indicating that miR-31a-5p was able to regulate endogenous RhoBTB1 expression levels in NRVMs. Moreover, qRT-PCR demonstrated that miR-31a-5p did not regulate the expression of other RhoBTBs, including RhoBTB2 and RhoBTB3, at least at the mRNA level (Figure 3d). Next, we sought to establish a mechanistic link between miR-31a-5p and its target gene RhoBTB1. RhoBTB1 knockdown mediated by the RhoBTB1 siRNA led to an increase in EdU incorporation into NRVMs (Figure 3e and f). Co-transfection with the RhoBTB1 siRNA and miR-31a-5p mimic did not exert an additive effect on the increase in EdU incorporation (Figure 3f). Moreover, co-transfection with the RhoBTB1 siRNA and miR-31a-5p inhibitor completely reversed the suppressive effect of the miR-31a-5p inhibitor on the EdU incorporation rate of NRVMs (Figure 3g).

RhoBTB1 is a target gene of miR-31a-5p. (a) Targetscan and luciferase assays showed RhoBTB1 as a direct target of miR-31a-5p. n=6 per group. (b, c) Western blot showed that miR-31a-5p endogenously negatively regulated RhoBTB1 in neonatal rat ventricular cardiomyocytes (NRVMs). n=3 per group. (d) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis demonstrated that miR-31a-3p did not regulate expression of RhoBTB2 and RhoBTB3 in NRVMs, at least at the mRNA level. n=6 per group. (e) qRT-PCR analysis of RhoBTB1 in cells transfected with its siRNA. n=3 per group. (f) Immunohistochemical stainings for sarcomeric α-actinin and 5-ethynyl-2-deoxyuridine (EdU) staining showed that RhoBTB1 knockdown induced an increase in EdU incorporation, whereas co-transfection with the RhoBTB1 siRNA and miR-31a-5p mimic did not exert an additive effect. At least 2000 cells were quantified in each group. Scale bar: 100 μm. (g) Immunohistochemical stainings for sarcomeric α-actinin and EdU staining showed that RhoBTB1 knockdown induced an increase in EdU incorporation, whereas co-transfection with the RhoBTB1 siRNA and miR-31a-5p inhibitor completely reversed the suppressive effect of the miR-31a-5p inhibitor on the EdU incorporation rate of NRVMs. At least 2000 cells were quantified in each group. Scale bar: 100 μm. *P<0.05, **P<0.01, ***P<0.001 versus respective control.
Taken together, these results demonstrate that RhoBTB1 is responsible for the pro-proliferative effect of miR-31a-5p in NRVMs.
miR-31a-5p is required for postnatal cardiomyocyte proliferation in vivo
To further explore whether miR-31a-5p is required for postnatal cardiomyocyte proliferation in vivo, neonatal rats were injected intraperitoneally with the miR-31a-5p antagomir or scrambled controls for three consecutive days after birth to prevent the increase in miR-31a-5p observed in the postnatal heart (Figure 4a). Inhibition of miR-31a-5p decreased cardiomyocyte proliferation, as evidenced by reduced PCNA expression, EdU incorporation and Ki-67 or phospho-histone-H3 (pHH3) staining in cardiomyocytes (Figure 4b–e). Importantly, as expected, miR-31a-5p downregulation was paralleled by an upregulation of its downstream target RhoBTB1 (Figure 4e).

miR-31a-5p is required for postnatal cardiomyocyte proliferation in vivo. (a) miR-31a-5p antagomirs significantly decreased miR-31a-5p expression in hearts. n=6 per group. (b–d) Antagonizing miR-31a-5p decreased cardiomyocyte proliferation, as shown by 5-ethynyl-2-deoxyuridine (EdU) incorporation (b) and Ki-67 (c) or phospho-histone-H3 (pHH3) staining (d) in cardiomyocytes. At least 2000 cells were quantified in each group. Scale bar: 50 μm. (e) Western blot showed that antagonizing miR-31a-5p reduced proliferating cell nuclear antigen (PCNA) expression, which was paralleled by an upregulation in its downstream target RhoBTB1. n=3 per group.
Collectively, these data indicate that miR-31a-5p is required for postnatal cardiomyocyte proliferation in vivo.
Discussion
Endogenous cardiomyocytes have been increasingly recognized as a source of new cardiomyocytes, which indicates the possibility of stimulating endogenous cardiomyocyte proliferation for myocardial repair and regeneration.1, 19, 20 Interestingly, the postnatal mammalian heart retains a regenerative capacity that is lost within 7 days, coinciding with the time when cardiomyocytes withdraw from the cell cycle after birth.16 Exploring factors promoting cardiomyocyte proliferation may help develop novel therapeutic strategies for enhancing cardiomyocyte proliferation following cardiac ischemic injury or heart failure.21
Few miRNAs have been reported to control postnatal cardiomyocyte proliferation.8 A previous miRNA array study using ventricles from mice at 1 day and 10 days of age found that the miR-15 family, especially miR-195, regulates the postnatal mitotic arrest of cardiomyocytes.16 Knockdown of the miR-15 family in neonatal mice increased the number of mitotic cardiomyocytes, indicating that the miR-15 family is a negative regulator of postnatal cardiomyocyte proliferation.16 Another study compared the expression levels of miRNAs in purified neonatal and adult cardiomyocyte, and found increase in miR-29a, miR-30a and miR-141 in adult cardiomyocytes.17 Treatment of neonatal cardiomyocytes with miR-29a, miR-30a or miR-141 inhibitors leads to more cycling cardiomyocytes.17 Similarly, comparisons of miRNA expression patterns in rat cardiomyocytes between postnatal day 2 (P2) and 4 weeks of age (P4W)5 showed that 95 miRNAs were differentially expressed in P4W cardiomyocytes. Overexpression of miR-29a suppressed the proliferation of the H9C2 cell line, whereas miR-29a inhibition increased it.5 Although promising, these studies used either whole-heart preparations or cultured neonatal cardiomyocytes at late time points (4 weeks or adult).5, 16, 17 Here, we took advantage of miRNA arrays to compare miRNA profiles in cardiomyocytes between P0 and P10 followed by a qRT-PCR validation and functional screening and found that miR-31a-5p was able to increase NRVM proliferation. miR-31a-5p expression was increased in cardiomyocytes isolated at P10, which is surprising and contrary to our initial hypothesis. We consider the upregulation of miR-31a-5p in P10 cardiomyocytes as a compensatory mechanism of the cardiomyocytes in response to exiting the cell cycle. However, determining whether miR-31a-5p is able to promote mature cardiomyocyte proliferation and whether RhoBTB1 is a target of miR-31a-5p in mature cardiomyocytes is highly needed. We cannot fully exclude the possibility that miR-31a-5p does not have an effect on mature cardiomyocyte proliferation.
In fact, relatively little is known about the role of miR-31a-5p (also known as miR-31) in the heart. miR-31a-5p has previously been confirmed as a negative modulator of the cardiac fibrogenic epithelial-to-mesenchymal transition of epicardial mesothelial cells.22 The atrial-specific increase in miR-31a-5p in human atrial fibrillation is a key factor mediating atrial loss of dystrophin and neuronal nitric oxide synthase.23 In addition, miR-31a-5p has been reported to inhibit the proliferation of nasopharyngeal carcinoma cells, serous epithelial ovarian cancer cells and glioblastoma multiform cells.24, 25, 26 However, miR-31a-5p has been demonstrated to promote the proliferation of colon cancer cells.27 These data indicate the complex roles of miR-31a-5p in cellular proliferation and support a tissue- and cell-based specific role of miR-31a-5p. Here, we first report a novel role of miR-31a-5p in controlling postnatal cardiomyocyte proliferation, which is supported by the fact that miR-31a-5p promotes NRVM proliferation, whereas its inhibition has the inverse effect in vitro. Furthermore, antagonizing miR-31a-5p is able to decrease cardiomyocyte proliferation in vivo. This is the first miRNA identified from postnatal heart growth that can promote NRVM proliferation.
RhoBTB proteins, including RhoBTB1, RhoBTB2 and RhoBTB3, are a subfamily of the Rho small GTPases, which are characterized by a modular organization and a conserved C-terminal region participating in signal transduction cascades.28 Relatively little is known about the function of RhoBTB1. RhoBTB1 is a tumor suppressor gene, located at the allelic loss region 10q21 in head and neck cancer.29 RhoBTB1 has also been identified as a target gene of miR-31 in human colon cancer.27 Interestingly, the role of RhoBTB1 in cardiac pathologies is completely unexplored to date. Here, we found that knockdown of RhoBTB1 could promote cardiomyocyte proliferation and was responsible for the pro-proliferative effect of miR-31a-5p in cardiomyocytes. Importantly, miR-31a-5p downregulation was paralleled with an upregulation in RhoBTB1 expression both in vitro and in vivo. It would also be interesting to further determine the in vivo roles for RhoBTB1 in the future. Nevertheless, the present study suggests that RhoBTB1 is a target gene of miR-31a-5p in cardiomyocytes and implicates potential roles of RhoBTB1 in the heart that deserve to be explored in the future.
Interestingly, the suppressive effect of miR-31a-5p inhibition in cardiomyocyte proliferation was also confirmed by the result that antagonizing miR-31a-5p decreased expression of cardiomyocyte proliferation markers in vivo. As it is beyond the scope of the present work, we did not examine whether miR-31a-5p functionally contributed to neonatal and adult mammalian cardiac regeneration or whether increasing miR-31a-5p protected against cardiac ischemic injury. Previous studies by others have indicated that miRNAs regulating the postnatal mitotic arrest of cardiomyocytes, such as the miR-15 family, may be modulators of heart regeneration, and their inhibition may protect against cardiac ischemic injury.30 Thus, the functional roles of miR-31a-5p in cardiac repair and regeneration under pathological conditions need to be clarified in the future.
In conclusion, miR-31a-5p controls postnatal cardiomyocyte proliferation by targeting RhoBTB1, and enhancing miR-31a-5p might be a novel strategy for promoting cardiac regeneration.
References
Rosenzweig A . Medicine: cardiac regeneration. Science 2012; 338: 1549–1550.
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN et al. Transient regenerative potential of the neonatal mouse heart. Science 2011; 331: 1078–1080.
Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA 2013; 110: 1446–1451.
Zacchigna S, Giacca M . Extra- and intracellular factors regulating cardiomyocyte proliferation in postnatal life. Cardiovasc Res 2014; 102: 312–320.
Cao X, Wang J, Wang Z, Du J, Yuan X, Huang W et al. Microrna profiling during rat ventricular maturation: a role for mir-29a in regulating cardiomyocyte cell cycle re-entry. FEBS Lett 2013; 587: 1548–1555.
Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013; 497: 249–253.
Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S et al. Functional screening identifies mirnas inducing cardiac regeneration. Nature 2012; 492: 376–381.
Piccoli MT, Gupta SK, Thum T . Noncoding rnas as regulators of cardiomyocyte proliferation and death. J Mol Cell Cardiol 2015; 89: 59–67.
Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T et al. miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia- reperfusion injury. Theranostics 2017; 7: 664–676.
De Rosa S, Curcio A, Indolfi C . Emerging role of micrornas in cardiovascular diseases. Circ J 2014; 78: 567–575.
Liu N, Olson EN . Microrna regulatory networks in cardiovascular development. Dev Cell 2010; 18: 510–525.
Thum T, Condorelli G . Long noncoding rnas and micrornas in cardiovascular pathophysiology. Circ Res 2015; 116: 751–762.
Olson EN . Micrornas as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med 2014; 6: 239ps233.
Boon RA, Dimmeler S . Micrornas in myocardial infarction. Nat Rev Cardiol 2015; 12: 135–142.
Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics 2016; 6: 2068–2083.
Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ et al. Mir-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res 2011; 109: 670–679.
Zhang Y, Matsushita N, Eigler T, Marban E . Targeted microrna interference promotes postnatal cardiac cell cycle re-entry. J Regen Med 2013; 2: 2.
Paradis AN, Gay MS, Wilson CG, Zhang L . Newborn hypoxia/anoxia inhibits cardiomyocyte proliferation and decreases cardiomyocyte endowment in the developing heart: role of endothelin-1. PLoS ONE 2015; 10: e0116600.
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013; 493: 433–436.
Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015; 523: 226–230.
Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014; 157: 565–579.
Bronnum H, Andersen DC, Schneider M, Nossent AY, Nielsen SB, Sheikh SP . Islet-1 is a dual regulator of fibrogenic epithelial-to-mesenchymal transition in epicardial mesothelial cells. Exp Cell Res 2013; 319: 424–435.
Reilly S, Liu X, Carnicer R, Rajakumar T, Sayeed R, Krasopoulos G et al. Evaluation of the role of mir-31-dependent reduction in dystrophin and nnos on atrial-fibrillation-induced electrical remodelling in man. Lancet 2015; 385 (Suppl 1): S82.
Cheung CC, Chung GT, Lun SW, To KF, Choy KW, Lau KM et al. Mir-31 is consistently inactivated in ebv-associated nasopharyngeal carcinoma and contributes to its tumorigenesis. Mol Cancer 2014; 13: 184.
Ibrahim FF, Jamal R, Syafruddin SE, Ab Mutalib NS, Saidin S, MdZin RR et al. Microrna-200c and microrna-31 regulate proliferation, colony formation, migration and invasion in serous ovarian cancer. J Ovarian Res 2015; 8: 56.
Zhou RJ, Xu XY, Liu BX, Dai WZ, Cai MQ, Bai CF et al. Growth-inhibitory and chemosensitizing effects of microrna-31 in human glioblastoma multiforme cells. Int J Mol Med 2015; 36: 1159–1164.
Xu RS, Wu XD, Zhang SQ, Li CF, Yang L, Li DD et al. The tumor suppressor gene rhobtb1 is a novel target of mir-31 in human colon cancer. Int J Oncol 2013; 42: 676–682.
Berthold J, Schenkova K, Rivero F . Rho gtpases of the rhobtb subfamily and tumorigenesis. Acta Pharmacol Sin 2008; 29: 285–295.
Beder LB, Gunduz M, Ouchida M, Gunduz E, Sakai A, Fukushima K et al. Identification of a candidate tumor suppressor gene rhobtb1 located at a novel allelic loss region 10q21 in head and neck cancer. J Cancer Res Clin Oncol 2006; 132: 19–27.
Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D et al. Regulation of neonatal and adult mammalian heart regeneration by the mir-15 family. Proc Natl Acad Sci USA 2013; 110: 187–192.
Acknowledgements
This work was supported by Innovation Program of Shanghai Municipal Education Commission, grants from the National Natural Science Foundation of China (91639101 and 81570362 to JX, 81370332 and 81170201 to XL, 81400647 to YB), the grant from Science and Technology Commission of Shanghai Municipality (17010500100), the development fund for Shanghai talents (to JX), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD20102013 to XL), the Netherlands Cardiovascular Research Initiative (CVON): the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science (to JPGS), the Romanian National Authority for Scientific Research, CNCS—UEFISCDI, projects number 82/2012 and 194/2014 for DC. Dr SD is supported by a grant from NHLBI (R01 HL122547). Dr XK is a Fellow and Dr XL is an Associate Fellow of the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Xiao, J., Liu, H., Cretoiu, D. et al. miR-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp Mol Med 49, e386 (2017). https://doi.org/10.1038/emm.2017.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.150
This article is cited by
-
SIRT3-dependent mitochondrial redox homeostasis mitigates CHK1 inhibition combined with gemcitabine treatment induced cardiotoxicity in hiPSC-CMs and mice
Archives of Toxicology (2023)
-
miRNA in cardiac development and regeneration
Cell Regeneration (2021)
-
A miRNA’s insight into the regenerating heart: a concise descriptive analysis
Heart Failure Reviews (2020)
-
Non-coding RNAs: emerging players in cardiomyocyte proliferation and cardiac regeneration
Basic Research in Cardiology (2020)
-
Cardiomyocyte Proliferation for Therapeutic Regeneration
Current Cardiology Reports (2018)
