
Abstract
Basal autophagy plays a critical role in maintaining cellular homeostasis and genomic integrity by degrading aged or malfunctioning organelles and damaged or misfolded proteins. However, autophagy also plays a complicated role in tumorigenesis and treatment responsiveness. It can be tumor-suppressing during the early stages of tumorigenesis (i.e., it is an anti-tumor mechanism), as reduced autophagy is found in tumor cells and may be associated with malignant transformation. In this case, induction of autophagy would seem to be beneficial for cancer prevention. In established tumors, however, autophagy can be tumor-promoting (i.e., it is a pro-tumor mechanism), and cancer cells can use enhanced autophagy to survive under metabolic and therapeutic stress. The pharmacological and/or genetic inhibition of autophagy was recently shown to sensitize cancer cells to the lethal effects of various cancer therapies, including chemotherapy, radiotherapy and targeted therapies, suggesting that suppression of the autophagic pathway may represent a valuable sensitizing strategy for cancer treatments. In contrast, excessive stimulation of autophagy may also provide a therapeutic strategy for treating resistant cancer cells having high apoptotic thresholds. In order for us to develop successful autophagy-modulating strategies against cancer, we need to better understand how the roles of autophagy differ depending on the tumor stage, cell type and/or genetic factors, and we need to determine how specific pathways of autophagy are activated or inhibited by the various anti-cancer therapies.
Similar content being viewed by others
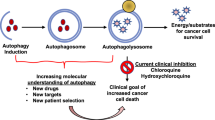


Introduction
Autophagy is a self-digestive process that ensures the lysosomal degradation of superfluous or damaged organelles and misfolded proteins (Levine and Klionsky, 2004). Basal autophagy helps maintain homeostasis by contributing to protein and organelle turnover, while additional autophagy is induced in stressed cells as a cell-survival mechanism. Autophagic defects have been implicated in various diseases and health states, including neurodegeneration, aging, infection, myopathy, Crohn's disease and cancer (Levine and Kroemer, 2008). However, the role of autophagy in cancer is quite complicated and still somewhat controversial; it appears to be tumor suppressive during cancer development, but contributes to tumor cell survival during cancer progression (Rouschop and Wouters, 2009). Furthermore, tumor cells can use autophagy to resist various anti-cancer therapies (Chen and Karantza-Wadsworth, 2009). Cancer cells experience higher metabolic demands and stresses than normal cells, due to their rapid proliferation and altered glycolytic metabolism (White and DiPaola, 2009), and thus may depend more heavily on autophagy for survival (Amaravadi et al., 2011). In many experimental settings, the inhibition of autophagy has been shown to enhance the therapeutic benefits of various cancer therapies (Chen and Karantza-Wadsworth, 2009; Chen et al., 2010). However, strategies to induce autophagic cell death have also shown promise as a means for killing certain types of cancer cells with high apoptotic thresholds (Gozuacik and Kimchi, 2007). Although we still have much to learn about the regulation of autophagy in cancer, it appears to provide a promising target for cancer treatment. This review examines the multiple roles of autophagy as a novel target for anticancer therapy and assesses the current efforts and remaining tasks in the development of autophagy-modulating strategies against cancer.
Roles of autophagy in cancer
Autophagy as a tumor-suppressing mechanism
Deficiencies in autophagy lead to the accumulation of damaged macromolecules and organelles (particularly mitochondria), subsequently inducing oxidative stress, DNA damage and chromatin instability (Levin and Klionsky, 2004; Chen and Karantza, 2011). Thus, autophagic defects are ultimately associated with the accumulation of oncogenic mutations and increased tumor susceptibility (Karanta-Wadsworth et al., 2007; Mathew et al., 2007). Some of the most important evidence for the role of autophagy in tumor suppression comes from studies on the Bcl-2-interacting protein, Beclin 1 (BECN1, also called ATG6). For example, monoallelic deletion of Beclin 1 has been frequently observed in human breast (Futreal et al., 1992), ovarian (Aita et al., 1999) and prostate cancers (Gao et al., 1995), and mice with a disrupted Beclin 1 gene had a higher incidence of lymphoma, lung cancer and liver cancer (Qu et al., 2003; Yue et al., 2003). Constitutive activation of the PI3K-Akt-mTOR axis, a common characteristic of cancer (LoPiccolo et al., 2008), is known to suppress autophagy (Janku et al., 2011), while promoting tumor cell growth, proliferation and survival (Martelli et al., 2007) (Figure 1). A number of tumor suppressor proteins have been shown to promote autophagy, including Atg4c (Mariño et al., 2007), Baxinteracting factor-1 (Bif-1) (Takahashi et al., 2007), BH3-only proteins (Maiuri et al., 2007), DAPkinase (Bialik and Kimchi, 2010), LKB1 (Liang et al., 2007), ultraviolet radiation resistance-associated gene (UVRAG) (Liang et al., 2006), PTEN (Arico et al., 2001), TSC (Zhou et al., 2009), nuclear p53 (Maiuri et al., 2010) and AMPK (Luo et al., 2010). Furthermore, autophagy has been shown to reduce intratumoral necrosis and local inflammation (Degenhardt et al., 2006). Collectively, these results support the contention that autophagy plays a tumor-suppressing role in cancer, and suggest that reduced autophagic activity may constitute a hallmark of cancer (Hanahan and Weinberg, 2011).

Oncogenes and tumor suppressors associated with the regulation of autophagy. Tumor suppressors (blue) except for cytoplasmic p53 are among the factors positively regulating autophagy, whereas oncogene products (pink) inhibit autophagy. Growth factor signaling activates the PI3K/Akt/mTOR axis resulting in autophagy inhibition. In contrast, class III PI3K activates autophagy. Low cellular energy levels with increased AMP/ATP ratio activate the LKB1-AMPK-mTOR pathway to also upregulate autophagy. p53 exhibits complex autophagy regulation, as nuclear p53 activated by genotoxic or oncogenic stress positively regulates autophagy by inhibiting mTOR in an activated AMPK- and TSC1/TSC2-dependent manner, whereas cytoplasmic p53 can suppress autophagy.
Autophagy as a pro-survival and resistance mechanism
In established cancer cells, metabolic stress (which arises as a result of insufficient nutrient or oxygen supplies and/or the increased energetic demands of rapidly dividing tumor cells) induces autophagy as the cells seek an alternative source of energy and metabolites (Onodera and Oshumi, 2005; Degenhardt et al., 2006; Jones and Thompson, 2009; Rosenfeldt and Ryan, 2009; Rabinowitz and White, 2010). Furthermore, autophagy may also be induced as an adaptive cellular response to various cancer therapies, leading to chemoresistance and cancer cell survival (Chen and Karantza-Wadsworth, 2009; White and DiPaola, 2009). A number of studies in various cancers have examined the inhibition of autophagy through pharmacological means, such as treatment with 3-methyladenine (3-MA, a PI3K III inhibitor), bafilomycin A (a specific inhibitor of vacuolar-type H+-ATPase), chloroquine (CQ) or hydroxychloroquine (HCQ) (lysosomotropic agents that impair fusion between autophagosomes and lysosomes), or by knockdown of autophagy-related genes, such as ATG5, ATG6 and ATG7. Table 1 summarizes preclinical studies supporting autophagy inhibition as an anticancer strategy. Notably, these inhibitions sensitized cancer cells to a wide range of therapeutic modalities, including genotoxic chemo- and radio-therapy, hormonal therapy and receptor tyrosine kinase inhibition (Chen et al., 2010). In this respect, the inhibition of autophagy appears to represent a major therapeutic means for sensitizing cells to various anti-cancer therapies.
Autophagy as a pro-death mechanism: autophagic cell death
Under extreme stress, tumor cells with apoptosis-defective genetic backgrounds die via other mechanisms (Kondo and Kondo, 2006; Gozuacik and Kimchi, 2007). Sustained autophagic activation, which leads to turnover of proteins and organelles beyond a survival threshold, can kill some cancer cells with high apoptotic thresholds, thus enhancing treatment efficacy (Yang et al., 2011). In this case, therefore, cell death via autophagy could provide an alternative therapeutic strategy (Gozuacik and Kimchi, 2007; Eisenberg-Lerner et al., 2009).
However, the concept of "autophagic cell death", also known as type II programmed cell death (Gozuacik and Kimchi, 2004), has been the subject of some controversy. It was initially regarded as a cell death mode that included the presence of autophagosomes (Schweichel and Merker, 1973). In many cases, however, autophagy is presumably activated by dying cells as part of an unsuccessful effort to cope with stress (i.e., as a pro-survival mechanism) (Boya et al., 2005). Increases in autophagic markers (e.g., autophagosome accumulation and up-regulation of the LC3 II form) in dying cells following exposure to chemotherapy or molecular targeted therapeutics do not necessarily indicate increases in autophagic flux (Mizushima et al., 2010). Instead, inefficient fusion between autophagosomes and lysosomes or reduced lysosomal degradation might lead to massive accumulation of autophagosomes. In this case, inhibition of autophagy would accelerate cell death rather than preventing it (Boya et al., 2005). Since the term "autophagic cell death" is highly prone to misinterpretation from a purely morphological perspective, the Nomenclature Committee on Cell Death very recently suggested that the term should only be used to indicate a cell death that is mediated by autophagy, as assessed based on biochemical and functional considerations (Galluzzi et al., 2012). In other words, "autophagic cell death" should be a cell death mode that is suppressed by inhibition of autophagy by chemicals (e.g., 3-MA) and/or genetic means, such as gene knockout/mutation or RNAi targeting of essential autophagic modulators, such as AMBRA1, ATG5, ATG12 or Beclin 1 (Galluzzi et al., 2012). Recently, Shen and Codogno (2011) also defined "autophagic cell death" as a form of programmed cell death in which autophagy per se serves as a cell death mechanism, in that it is cell death by autophagy, not cell death with autophagy. They proposed that autophagic cell death should meet the following criteria: 1) the cell death occurs without apoptosis; 2) there is an increase of the autophagic flux, not just the autophagic markers, in dying cells; and 3) suppression of autophagy via both pharmacological inhibitors and genetic approaches can rescue or prevent the cell death. Such autophagic cell death has been reported during the in vivo development of D. melanogaster (Berry and Baehrecke, 2007; Denton et al., 2009) and in hippocampal neural stem cells of adult rat brain following insulin withdrawal (Yu et al., 2008). However, the in vivo evidence of autophagic cell death in mammals has been relatively limited to date.
Targeting autophagy for cancer therapy
Induction of autophagy for cancer prevention
Various dietary phytochemicals, including β-carotene, lycopene, lutein, quercetin, resveratrol, curcumin and epigallocatechin-3-gallate (EGCG) have demonstrated chemopreventive activities in many preclinical and clinical studies (Davis 2007; Pan and Ho, 2008). Their antioxidant, anti-inflammatory, and pro-apoptotic activities appear to be important for preventing, suppressing, or reversing the development of carcinogenesis (Tan et al., 2011). Both in vitro and animal studies have demonstrated that a number of phytochemicals (curcumin, resveratrol, EGCG, sulforaphane and silibinin) show preferential cytotoxicity to malignant cancer cells over normal cells, suggesting that they might safely be used for both cancer chemoprevention and cancer therapy (Nair et al., 2007; Mann et al., 2009). Interestingly, a number of dietary phytochemicals, including quercetin, apigenin, genistein, hesperetin, and luteolin were recently shown to induce autophagy in both normal cells and several cancer cell types (Singletary and Milner, 2008; Hannigan and Gorski, 2009). Furthermore, pharmacological activation of autophagy by drugs, such as rapamycin analogs (mTOR inhibitors, (Harrison et al., 2009)), class I PI3K inhibitors (Levine and Kroemer, 2008) and metformin (an AMPK activator, (Buzzai et al., 2007)) were demonstrated to inhibit malignant transformation (Evans et al., 2005). The possibility of preventing cancer by activating autophagy is very intriguing. However, it is not yet clear whether the enhancement of autophagy is a potentially critical mechanism for the chemopreventive and/or chemotherapeutic actions of these agents. Future work will be required to determine whether the up-regulation of autophagy is a safe and effective approach for cancer prevention.
Inhibiting autophagy to enhance the efficacy of anticancer therapies
Despite recent advances in cancer treatment, many tumors still exhibit unsatisfactory responses to chemotherapy and/or radiation, either recurring or continuing to grow after treatment (Chen et al., 2010). Autophagy is commonly up-regulated in both tumor and normal cells exposed to cancer therapies, but the greater reliance of tumor cells (versus normal cells) on the cytoprotective effects of autophagy provides a novel therapeutic opportunity (White and DiPaola, 2009). Indeed, autophagy is induced as a pro-survival strategy in human cancer cells treated with HDAC inhibitors (Carew et al., 2007), arsenic trioxide (Smith et al., 2010), TNF-α (Moussay et al., 2011), IFN-γ (Ní Cheallaigh et al., 2011), rapamycin (Fan et al., 2010), and antiestrogen hormonal therapy (Qadir et al., 2008), suggesting that the inhibition of autophagy could sensitize cancer cells to these therapies. Consistent with this notion, siRNA-mediated suppression of ATG5, ATG6, or ATG7 as well as co-treatment with 3-MA or bafilomycin A1 sensitized various cancer cells to various anti-cancer drugs or therapies (Chen et al., 2010).
CQ derivatives act as lysosomotropic agents by increasing pH and inhibiting autophagosomal maturation (White and DiPaola, 2009). These oral drugs cross the blood-brain barrier (Amaravadi et al., 2011) and have been prescribed for malaria (O'Neill et al., 1998), rheumatoid arthritis (Kremer, 2001), and HIV (Romanelli et al., 2004), Recently, CQ and HCQ were shown to enhance the cytotoxic effects of chemotherapy or standard cancer therapy in vitro (Amaravadi et al., 2011). In addition, combined treatments with CQ or HCQ plus metabolic stressors (e.g., an angiogenesis inhibitor or 2-deoxyglucose) (Ramakrishnan et al., 2007; Dipaola et al., 2008) or targeted therapeutic drugs (e.g., imatinib, an inhibitor of Bcr-Abl) have been found to potentiate cell death (Shingu et al., 2009; Calabretta and Salomoni, 2011). HCQ is less toxic than CQ (Gunja et al., 2009) but has similar ability to induce autophagy (Amaravadi et al., 2011). Thus, multiple clinical trials are currently assessing the effects of combined treatments with various anti-cancer drugs plus HCQ for patients with various refractory malignancies (http://clinicaltrials.gov) (White and DiPaola, 2009; Levy and Thorburn, 2011). Since the pharmacological modulation of autophagy appears to enhance the efficacy of current anticancer regimens, these new combinatorial strategies may hopefully contribute to cancer eradication in the future.
Induction of autophagic cell death as a therapeutic strategy
Since defects in apoptosis are often observed in many solid tumor cells and can increase the resistance of tumor cells to various conventional cancer therapies, the targeting of alternative cell death pathways is an attractive strategy for improving anti-tumor therapy (Schleicher et al., 2010). Thus, the induction of cell death by autophagy may serve as a novel therapeutic strategy for eliminating cancer cells, especially those with high thresholds to apoptosis (Gozuacik and Kimchi, 2007). Although several agents, including arsenic trioxide (Kanzawa et al., 2003) and vitamin D analog EB1089 (Høyer-Hansen et al., 2005), were previously reported to induce autophagic cell death in cancer cells in vitro, in these cases, unfortunately, autophagic cell death was often determined based on morphological characteristics, meaning that some may not represent true autophagic cell death (Kroemer and Levine, 2008). However, several other reports, have demonstrated concrete examples of autophagic cell death in response to certain agents, further showing that the inhibition or knockdown of essential regulators of autophagy could prolong cell survival. Some cancer cells (especially those lacking essential apoptotic modulators, such as BAX, BAK or caspases) were found to show autophagic cell death in vitro when treated with certain chemotherapeutic agents, such as etoposide, fenretinide and dexamethasone (Shimizu et al., 2004; Fazi et al., 2008; Grander et al., 2009; Laane et al., 2009). Certain phytochemicals, including fisetin and resveratrol, have also been shown to induce autophagic cell death in certain types of cancer cells (Puissant et al., 2010; Suh et al., 2010). Furthermore, we recently reported that sodium selenite induces mitophagic cell death (cell death by excessive mitophagy) selectively in malignant glioma cells, but not in normal astrocytes (Kim et al., 2007; Kim and Choi, 2008), suggesting that selenite may prove useful for cancer therapy via a mitochondria-selective type of autophagic cell death. Table 2 lists other examples of autophagic cell death in cancer cells. Autophagic cell death seems to be induced in a cell type-, genetic background-, and stimulus-specific manner. Future studies will be needed to clarify whether the induction of autophagic cancer cell death will prove useful in the clinic.
Remaining tasks
Requirement for a novel and reliable method for measuring dynamic autophagic processes
Our current understanding of autophagy is undoubtedly incomplete. While we have a fairly good understanding of the regulatory mechanisms governing the early steps of autophagy, including autophagosome formation (initiation, nucleation and elongation) (Mizushima and Yoshimori, 2007), those governing the later steps of autophagy, such as autophagosomal maturation and lysosomal degradation, are less well understood. The widely used LC3-based autophagic assay primarily measures the early phases of autophagic activity, which control autophagosome formation (Kabeya et al., 2000). Recently, several different assays have been used to monitor autophagic flux, including the LC3 turnover assay, measurements of the total levels of autophagic substrates (e.g., LC3, p62 and GFP-LC3) (Mizushima and Yoshimori, 2007), analysis of the mRFP-GFP-LC3 color change (Kimura et al., 2007), and measurement of free GFP generated from GFP-LC3 (Hosokawa et al., 2006). However, the utilities and limitations of these assays may vary somewhat in different cell types and experimental contexts (Mizushima et al., 2010). For us to clearly understand the entire dynamic process of autophagy and identify therapeutic targets for its modulation, we need to develop new assays that can be used to examine autophagic activity all the way to the final lysosomal step. In addition, it would be useful to quantitatively measure activity changes among key autophagic effectors, and we should seek to generate standard autophagic assays suitable for routine clinical use. Currently, there are no available data on autophagic markers in biopsies obtained before and after treatment, which further indicates that we need new techniques for assessing autophagy in clinical samples, such as tumor biopsies and peripheral-blood mononuclear cells.
Rational therapeutic design based on the functional status of autophagy in cancer cells
Although only some of the links between autophagy and cancer have been elucidated in detail, pharmacological modulation of autophagy based on its functional status in tumors may provide novel opportunities for cancer management. However, the complex roles of autophagy in tumorigenesis and treatment responses make it difficult to decipher how we should modulate autophagy for maximum therapeutic benefit, given that context- and cell type-specific approaches may be required. Chronically autophagy-deficient tumors may be particularly sensitive to genotoxic and/or metabolic stress-inducing anticancer agents, such as DNA-damaging and anti-angiogenic drugs, due to the absence of autophagy-mediated survival mechanism (Chen and Karantza, 2011). In autophagy-competent tumors, concurrent autophagic inhibition would be expected to increase the efficacy of many anticancer modalities (Amaravadi et al., 2011). However, in autophagy-competent tumor cells with particular genetic backgrounds (for example, those having high thresholds of apoptosis), sustained or overstimulation of autophagy can lead to cell death, and enhanced treatment efficacy (Buytaert et al., 2006; Yang et al., 2011). Therefore, gaining detailed information on the cellular functional status of autophagy (together with apoptosis) in certain cancer types may facilitate the rational design of therapeutic regimens via the modulation of autophagy (Table 3). Context-specific pharmacological modulation of autophagy thus holds great promise as a novel therapeutic approach against cancer.
Establishment of effective combinatorial therapeutic strategies using inhibitors of autophagy
Several agents that were previously identified as inducers of autophagy (mainly by LC3-based autophagic assays), including thapsigargin (Høyer-Hansen et al., 2007) and imatinib (Ertmer et al., 2007), were recently reported to inhibit the lysosomal steps (autophagosomal-lysosomal fusion or lysosomal degradation) in different experimental settings (Yogalingam and Pendergast, 2008; Ganley et al., 2011). Inhibitors of autophagy (e.g., HCQ) are already being used in clinical trials along with various chemotherapeutic agents. Thus, when seeking to modulate autophagy in order to increase treatment efficacy, we should first evaluate whether the potentially co-treated anti-cancer drug increases or inhibits the autophagic flux in the target cancers.
The abrogation of autophagy often tends to sensitize therapeutic agent-resistant cancer cells to various anti-cancer treatments (Chen et al., 2010). Notably, however, blocking autophagy at different steps (i.e., inhibiting the autophagosome-formation step with 3-MA versus inhibiting the autophagosome-lysosome fusion step with CQ or HCQ) can have different or even opposing outcomes. Temozolomide- or imatinib-induced cytotoxicity was attenuated by the inhibition of autophagy at an early stage, but augmented by inhibition of autophagy at a late stage (Kanzawa et al., 2004; Shingu et al., 2009). At present, we do not clearly understand how the inhibition of autophagy stage-specifically affects cytotoxicity. Comparative studies on the effects of various combined therapies with agents that inhibit different autophagic stages may contribute to the rational design of combined regimens against cancer.
Although autophagic manipulation by inhibitors such as CQ and HCQ is already being used in clinical trials, these agents are known to have additional effects, such as immunomodulation and the possible induction of DNA damage at higher doses (Chen and Karantza, 2011). Thus, the side-effects of these autophagic inhibitors (e.g., potential induction of genomic instability, and adverse effect on the central nervous and cardiovascular systems) may make them suitable for only certain clinical situations. Furthermore, when we target the autophagic system for anti-cancer efforts, we must carefully consider the multifaceted nature of autophagy and its diverse crosstalk with other biological processes, including metabolism and the cell death pathways.
Conclusion
The molecular mechanisms underlying the regulation of autophagy and the role of autophagy in cancer cells are not completely understood. However, the pharmacological modulation of autophagy appears to have significant clinical potential as a novel therapeutic strategy for cancer eradication. The induction of autophagy may be useful for cancer chemoprevention in normal cells or triggering an alternative cell death mechanism in certain cancer cells, especially those with compromised apoptotic functions. In addition, suppression of the autophagic pathway may be combined with conventional antitumor regimens to achieve increased efficacy, representing a valuable therapeutic strategy for radio- and chemosensitization. In the future, we should seek to identify novel biomarkers for assessing dynamic autophagic processes and to establish new methods to assess autophagy in clinical samples. These efforts will help us understand the complicated role of autophagy in cancer and facilitate the rational design of combinatorial strategies aimed at modulating autophagy. Furthermore, future efforts toward universally modulating autophagy for maximum therapeutic benefit should focus on elucidating the genetic and physiological conditions that determine the pro-survival or pro-death functions of autophagy.
Abbreviations
- 3-MA:
-
3-methyladenine
- AMBRA1:
-
activating molecule in Beclin-1-regulated autophagy protein 1
- AMPK:
-
5' AMP-activated protein kinase or AMP-activated protein kinase
- ATG:
-
autophagyrelated gene
- BFA:
-
bafilomycin A1
- CQ:
-
chloroquine
- DAPkinase:
-
death associated protein (DAP) kinase
- EGCG:
-
epigallocatechin-3-gallate
- HCQ:
-
hydroxychloroquine
- HDAC inhibitor:
-
histone deacetylases inhibitor
- mTOR:
-
mammalian target of rapamycin
- PTEN:
-
phosphatase and tensin homolog
- RNAi:
-
RNA interference
- TSC:
-
tuberous sclerosis complex
References
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B . loning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21 . Genomics 1999 ; 59 : 59 - 65
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB . Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma . J Clin Invest 2007 ; 117 : 326 - 336
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E . Principles and current strategies for targeting autophagy for cancer treatment . Clin Cancer Res 2011 ; 17 : 654 - 666
Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A . Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy . Cancer Res 2008 ; 68 : 1485 - 1494
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E . The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway . J Biol Chem 2001 ; 276 : 35243 - 35246
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, Van Etten RA, Donato N, Hunter A, Dinsdale D, Tirrò E, Vigneri P, Nicotera P, Dyer MJ, Holyoake T, Salomoni P, Calabretta B . Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells . J Clin Invest 2009 ; 119 : 1109 - 1123
Berry DL, Baehrecke EH . Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila . Cell 2007 ; 131 : 1137 - 1148
Bialik S, Kimchi A . Lethal weapons: DAP-kinase, autophagy and cell death: DAP-kinase regulates autophagy . Curr Opin Cell Biol 2010 ; 22 : 199 - 205
Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, Jiang Y, Singh SV . Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells . Cancer Res 2009 ; 69 : 3704 - 3712
Boya P, González-Polo RA, Casares N, Perferrini JL, Dessed P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G . Inhibition of macroautophagy triggers apoptosis . Mol Cell Biol 2005 ; 25 : 1025 - 1040
Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, Walker R, Hermann RS . Active cell death induced by theanti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy . Carcinogenesis 1996 ; 17 : 1595 - 1607
Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, Vandenheede JR, Heirman I, Grooten J, Agostinis P . Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy . FASEB J 2006 ; 20 : 756 - 758
Buzzai M, Jones RG, Amaravadi RK, Lum JJ, De Berardinis RJ, Zhao F, Viollet B, Thompson CB . Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth . Cancer Res 2007 ; 67 : 6745 - 6752
Calabretta B, Salomoni P . Inhibition of autophagy: a new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors . Leuk Lymphoma 2011 ; 52 : 54 - 59
Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL . Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance . Blood 2007 ; 110 : 313 - 322
Chen N, Karantza-Wadsworth V . Role and regulation of autophagy in cancer . Biochim Biophys Acta 2009 ; 1793 : 1516 - 1523
Chen N, Karantza V . Autophagy as a therapeutic target in cancer . Cancer Biol Ther 2011 ; 11 : 157 - 168
Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J . Autophagy is a therapeutic target in anticancer drug resistance . Biochim Biophys Acta 2010 ; 1806 : 220 - 229
Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, Agostinis P, Garmyn M . Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma . Int J Cancer 2010 ; 127 : 2790 - 2803
Crowley LC, Elzinga BM, O'Sullivan GC, McKenna SL . Autophagy induction by Bcr-Abl-expressing cells facilitates their recovery from a targeted or nontargeted treatment . Am J Hematol 2011 ; 86 : 38 - 47
Davis CD . Nutritional interactions: Credentialing of molecular targets for cancer prevention . Exp Biol Med (Maywood) 2007 ; 232 : 176 - 183
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, Nelson DA, Jin S, White E . Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis . Cancer Cell 2006 ; 10 : 51 - 64
Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumars S . Autophagy, not apoptosis, is essential for midgut cell death in Drosophila . Curr Biol 2009 ; 19 : 1741 - 1746
Dikic I, Johansen T, Kirkin V . Selective autophagy incancer development and therapy . Cancer Res 2010 ; 70 : 3431 - 3434
DiPaola RS, Dvorzhinski D, Thalasila A, Garikapaty V, Doram D, May M, Bray K, Mathew R, Beaudoin B, Karp C, Stein M, Foran DJ, White E . Therapeutic starvation and autophagy in prostate cancer: a new paradigm for targeting metabolism in cancer therapy . Prostate 2008 ; 68 : 1743 - 1752
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A . Life and death partners: apoptosis, autophagy and the cross-talk between them . Cell Death Differ 2009 ; 16 : 966 - 975
Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Durster J, Elsässer M, Schätzl HM . The anticancer drug imatinib induces cellular autophagy . Leukemia 2007 ; 21 : 936 - 942
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD . Metformin and reduced risk of cancer indiabetic patients . BMJ 2005 ; 330 : 1304 - 1305
Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, Shokat KM, Weiss WA . Akt and autophagy cooperate to promote survival of drug-resistant glioma . Sci Signal 2010 ; 3 : ra81 -
Fazi B, Bursch W, Fimia GM, Nardacci R, Piacentini M, Di Sano F, Piredda L . Fenretinide induces autophagic cell death in caspase-defective breast cancer cells . Autophagy 2008 ; 4 : 435 - 441
Fels DR, Ye J, Segan AT, Kridel SJ, Spiotto M, Olson M, Koong AC, Koumenis C . Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways . Cancer Res 2008 ; 68 : 9323 - 9330
Futreal PA, Söderkvist P, Marks JR, Iglehart JD, Cochran C, Barrett JC, Wiseman RW . Detection of frequent allelic loss on proximal chromosome 17q in sporadic breast carcinoma using microsatellite length polymorphisms . Cancer Res 1992 ; 52 : 2624 - 2627
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knigh RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G . Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012 . Cell Death Differ 2012 ; 19 : 107 - 120
Ganley IG, Wong PM, Gammoh N, Jiang X . Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest . Mol Cell 2011 ; 42 : 731 - 743
Gao X, Zacharek A, Salkowski A, Grignon DJ, Sakr W, Porter AT, Honn KV . Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer . Cancer Res 1995 ; 55 : 1002 - 1005
Gozuacik D, Kimchi A . Autophagy and cell death . Curr Top Dev Biol 2007 ; 78 : 217 - 245
Gozuacik D, Kimchi A . Autophagy as a cell death and tumor suppressor mechanism . Oncogene 2004 ; 23 : 2891 - 2906
Grander D, Kharaziha P, Laane E, Pokrovskaja K, Panaretakis T . Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies . Autophagy 2009 ; 5 : 1198 - 1200
Gunja N, Roberts D, McCoubrie D, Lamberth P, Jan A, Simes DC, Hackett P, Buckley NA . Survival after massive hydroxychloroquine overdose . Anaesth Intensive Care 2009 ; 37 : 130 - 133
Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, McMahon J, Taguchi T, Floris G, Debiec-Rychter M, Schöffski P, Trent JA, Debnath J, Rubin BP . Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST) . Proc Natl Acad Sci USA 2010 ; 107 : 14333 - 14338
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation . Cell 2011 ; 144 : 646 - 674
Hannigan AM, Gorski SM . Macroautophagy: the key ingredient to a healthy diet ? Autophagy 2009 ; 5 : 140 - 151
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA . Rapamycin fed late in life extends lifespan in genetically heterogeneous mice . Nature 2009 ; 460 : 392 - 395
Hoare M, Young AR, Narita M . Autophagy in cancer: Having your cake and eating it . Semin Cancer Biol 2011 ; 21 : 397 - 404
Hosokawa N, Hara Y, Mizushima N . Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size . FEBS Lett 2006 ; 580 : 2623 - 2629
Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M . Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death . Cell Death Differ 2005 ; 12 : 1297 - 1309
Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jäättelä M . Control of macro-autophagy by calcium, calmodulin-dependent kinase kinase-β,and Bcl-2 . Mol Cell 2007 ; 25 : 193 - 205
Janku F, McConkey DJ, Hong DS, Kurzrock R . Autophagy as a target for anticancer therapy . Nat Rev Clin Oncol 2011 ; 8 : 528 - 539
Jones RG, Thompson CB . Tumor suppressors and cell metabolism: a recipe for cancer growth . Genes Dev 2009 ; 23 : 537 - 548
Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, Neufeld TP . The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila . J Cell Biol 2008 ; 181 : 655 - 666
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Oshumi Y, Yoshimori T . LC3, a mammalian homologue of yeast Apg8p, Is localized in autophagosome membranes after processing . EMBO J 2000 ; 19 : 5720 - 5728
Kanematsu S, Uehara N, Miki H, Yoshizawa K, Kawanaka A, Yuri T, Tsubura A . Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells . Anticancer Res 2010 ; 30 : 3381 - 3390
Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I . Induction of autophagic cell death in malignant glioma cells by arsenic trioxide . Cancer Res 2003 ; 63 : 2103 - 2108
Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S . Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells . Cell Death Differ 2004 ; 11 : 448 - 457
Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E . Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis . Genes Dev 2007 ; 21 : 1621 - 1635
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, Naito M, Itoh M, Tomoda A . Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress . Int J Oncol 2011 ; 38 : 643 - 654
Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS . Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells . Cancer Res 2007 ; 67 : 6314 - 6324
Kim EH, Choi KS . A critical role of superoxide anion in selenite-induced mitophagic cell death . Autophagy 2008 ; 4 : 76 - 78
Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, Gandour-Edwards R, Chuang FY, Bold RJ, Kung HJ . Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis . Cancer Res 2009 ; 69 : 700 - 708
Kimura S, Noda T, Yoshimori T . Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3 . Autophagy 2007 ; 3 : 452 - 460
Kondo Y, Kondo S . Autophagy and cancer therapy . Autophagy 2006 ; 2 : 85 - 90
Kremer JM . Rational use of new and existing disease-modifying agents in rheumatoid arthritis . Ann Intern Med 2001 ; 134 : 695 - 706
Kroemer G, Levine B . Autophagic cell death: the story of a misnomer . Nat Rev Mol Cell Biol 2008 ; 9 : 1004 - 1010
Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J, Corcoran M, Björklund A-C, Hultenby K, Lundin J, Heyman M, Söderhäll S, Mazur J, Porwit A, Pandolfi PP, Zhivotovsky B, Panaretakis T, Grandér D . Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy . Cell Death Differ 2009 ; 16 : 1018 - 1029
Levine B, Klionsky DJ . Development of self-digestion:molecular mechanisms and biological functions of autophagy . Dev Cell 2004 ; 6 : 463 - 467
Levine B, Kroemer G . Autophagy in the pathogenesisof disease . Cell 2008 ; 132 : 27 - 42
Levy JM, Thorburn A . Targeting autophagy during cancer therapy to improve clinical outcomes . Pharmacol Ther 2011 ; 131 : 130 - 141
Li J, Hou N, Faried A, Tsutsumi S, Kuwano H . Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model . Eur J Cancer 2010 ; 46 : 1900 - 1909
Li M, Jiang X, Liu D, Na Y, Gao GF, Xi Z . Autophagy protects LNCaP cells under androgen deprivation conditions . Autophagy 2008 ; 4 : 54 - 60
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU . Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG . Nat Cell Biol 2006 ; 8 : 688 - 699
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB . The energy sensing LKB1-AMPK pathway regulates p27 (kip1) phosphorylation mediating the decision to enter autophagy or apoptosis . Nat Cell Biol 2007 ; 9 : 218 - 224
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B . Induction of autophagy and inhibition of tumorigenesis by beclin 1 . Nature 1999 ; 402 : 672 - 676
Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M . PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches . Cell Cycle 2009 ; 8 : 1352 - 1358
Ling YH, Aracil M, Zou Y, Yuan Z, Lu B, Jimeno J, Cuervo AM, Perez-Soler R . PM02734 (elisidepsin) induces caspase-independent cell death associated with features of autophagy, inhibition of the Akt/mTOR signaling pathway, and activation of death-associated protein kinase . Clin Cancer Res 2011 ; 17 : 5353 - 5366
Liu D, Yang Y, Liu Q, Wang J . Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells . Med Oncol 2011 ; 28 : 105 - 111
Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, Mikkelsen T, Brodie C . The induction of autophagy by γ-radiation contributes to the radioresistance of glioma stem cells . Int J Cancer 2009 ; 125 : 717 - 722
LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA . Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations . Drug Resist Updat 2008 ; 11 : 32 - 50
Luo Z, Zang M, Guo W . AMPK as a metabolic tumor suppressor: control of metabolism and cell growth . Future Oncol 2010 ; 6 : 457 - 470
Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G . BH3-onlyproteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) . Autophagy 2007 ; 3 : 374 - 376
Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G . Autophagy regulation by p53 . Curr Opin Cell Biol 2010 ; 22 : 181 - 185
Mann CD, Neal CP, Garcea G, Manson MM, Dennison AR, Berry DP . Phytochemicals as potential chemopreventive and chemotherapeutic agents in hepatocarcinogenesis . Eur J Cancer Prev 2009 ; 18 : 13 - 25
Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C . Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3 . J Biol Chem 2007 ; 282 : 18573 - 18583
Martelli AM, Tazzari PL, Evangelisti C, Chiarini F, Blalock WL, Billi AM, Manzoli L, McCubrey JA, Cocco L . Targeting the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Module for Acute Myelogenous Leukemia Therapy: From Bench to Bedside . Curr Med Chem 2007 ; 14 : 2009 - 2023
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E . Autophagy suppresses tumor progression by limiting chromosomal instability . Genes Dev 2007 ; 21 : 1367 - 1381
Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL . The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib . Cancer Res 2009 ; 69 : 4415 - 4423
Mizushima N, Yoshimori T . How to interpret LC3 immunoblotting . Autophagy 2007 ; 3 : 542 - 545
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research . Cell 2010 ; 140 : 313 - 326
Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N, Nazarov PV, Vallar L, Chouaib S, Berchem G, Janji B . The acquisition of resistance to TNFα in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray . Autophagy 2011 ; 7 : 760 - 770
Nair S, Li W, Kong AN . Natural dietary anti-cancer chemopreventive compounds: redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells . Acta Pharmacol Sin 2007 ; 28 : 459 - 472
Ní Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J . Autophagy in the immune response to tuberculosis: clinical perspectives . Clin Exp Immunol 2011 ; 164 : 291 - 300
Nishikawa T, Tsuno NH, Okaji Y, Shuno Y, Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K, Nagawa H . Inhibition of autophagy potentiates sulforaphane-induced apoptosis in human colon cancer cells . Ann Surg Oncol 2010 ; 17 : 592 - 602
O'Neill PM, Bray PG, Hawley SR, Ward SA, Park BK . 4-Aminoquinolines-past, present, and future: a chemical perspective . Pharmacol Ther 1998 ; 77 : 29 - 58
Onodera J, Ohsumi Y . Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation . J Biol Chem 2005 ; 280 : 31582 - 31586
Paglin S, Hollister T, Delohery T, Kackett N, McMahill M, Sphicas E, Domingo D, Yahalom J . A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles . Cancer Res 2001 ; 61 : 439 - 444
Pan MH, Ho CT . Chemopreventive effects of natural dietary compounds on cancer development . Chem Soc Rev 2008 ; 37 : 2558 - 2574
Park EJ, Choi KS, Kwon TK . β-Lapachone-induced reactive oxygen species (ROS) generation mediates autophagic cell death in glioma U87 MG cells . Chem Biol Interact 2011 ; 189 : 37 - 44
Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, Auberger P . Resveratrol promotes autophagic cell death in chronic myeogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation . Cancer Res 2010 ; 70 : 1042 - 1052
Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK . Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death . J Biol Chem 2005 ; 280 : 20722 - 20729
Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, Gorski SM . Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization . Breast Cancer Res Treat 2008 ; 112 : 389 - 403
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rose J, Eskelinen EL, Mizushima N, Oshumi Y, Cattoretti G, Levine B . Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene . J Clin Invest 2003 ; 112 : 1809 - 1820
Rabinowitz JD, White E . Autophagy and metabolism . Science 2010 ; 330 : 1344 - 1348
Ramakrishnan S, Nguyen TM, Subramanian IV, Kelekar A . Autophagy and angiogenesis inhibition . Autophagy 2007 ; 3 : 512 - 515
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-anchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC . Regulation of mammalian autophagy in physiology and pathophysiology . Physiol Rev 2010 ; 90 : 1383 - 1435
Romanelli F, Smith KM . Hoven AD. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity . Curr Pharm Des 2004 ; 10 : 2643 - 2648
Rosenfeldt MT, Ryan KM . The role of autophagy in tumour development and cancer therapy . Expert Rev Mol Med 2009 ; 11 : e36 -
Rouschop KM, Wouters BG . Regulation of autophagy through multiple independent hypoxic signaling pathways . Curr Mol Med 2009 ; 9 : 417 - 424
Salazar M, Carracedo A, Salanueva IJ, Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C, Torres S, García S, Nowak J, Fimia GM, Piacentini M, Cecconi F, Pandolfi PP, González-Feria L, Iovanna JL, Guzmán M, Boya P, Velasco G . Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells . J Clin Invest 2009 ; 119 : 1359 - 1372
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler K, Vogelstein B, Velculescu VE . High frequency of mutations of the PIK3CA gene in human cancers . Science 2004 ; 304 : 554 -
Schleicher SM, Moretti L, Varki V, Lu B . Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches . Drug Resist Updat 2010 ; 13 : 79 - 86
Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT . Autophagy facilitates the progression of ERα-positive breast cancer cells to antiestrogen resistance . Autophagy 2009 ; 5 : 400 - 403
Schweichel JU, Merker HJ . The morphology of various types of cell death in prenatal tissues . Teratology 1973 ; 7 : 253 - 266
Shen HM, Codogno P . Autophagic cell death:Loch Ness monster or endangered species ? Autophagy 2011 ; 7 : 457 - 465
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y . Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes . Nat Cell Biol 2004 ; 6 : 1221 - 1228
Shingu T, Fujiwara K, Bögler O, Akiyama Y, Moritake K, Shinojima N, Tamada Y, Yokoyama T, Kondo S . Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells . Int J Cancer 2009 ; 124 : 1060 - 1071
Singletary K, Milner J . Diet, autophagy, and cancer: a review . Cancer Epidemiol Biomarkers Prev 2008 ; 17 : 1596 - 1610
Smith DM, Patel S, Raffoul F, Haller E, Mills GB, Nanjundan M . Arsenic trioxide induces a beclin-1-independent autophagic pathway via modulation of SnoN/SkiL expression in ovarian carcinoma cells . Cell Death Differ 2010 ; 17 : 1867 - 1881
Suh Y, Afaq F, Khan N, Johnson JJ, Khusro FH, Mukhtar H . Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells . Carcinogenesis 2010 ; 31 : 1424 - 1433
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, Pledger WJ, Wang HG . Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis . Nat Cell Biol 2007 ; 9 : 1142 - 1151
Tan AC, Konczak I, Sze DM, Ramzan I . Molecular pathways for cancer chemoprevention by dietary phytochemicals . Nutr Cancer 2011 ; 63 : 495 - 505
Tiwari M, Bajpai VK, Sahasrabuddhe AA, Kumar A, Sinha RA, Behari S, Godbole MM . Inhibition of N-(4-hydroxyphenyl) retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells . Carcinogenesis 2008 ; 29 : 600 - 609
Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA . Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab . PLoS One 2009 ; 4 : e6251 -
Wang K, Liu R, Li J, Mao J, Lei Y, Wu J, Zeng J, Zhang T, Wu H, Chen L, Huang C, Wei Y . Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling . Autophagy 2011 ; 7 : 966 - 978
White E, DiPaola RS . The double-edged sword of autophagy modulation in cancer . Clin Cancer Res 2009 ; 15 : 5308 - 5316
Wu Z, Chang PC, Yang JC, Chu CY, Wang LY, Chen NT, Ma AH, Desai SJ, Lo SH, Evans CP, Lam KS, Kung HJ . Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors . Genes Cancer 2010 ; 1 : 40 - 49
Yang ZJ, Chee CE, Huang S, Sinicrope FA . The role of autophagy in cancer: therapeutic plications . Mol Cancer Ther 2011 ; 10 : 1533 - 1541
Yao KC, Komata T, Kondo Y, Kanazawa T, Kondo S, Germano IM . Molecular response of human glioblastoma multiforme cells to ionizing radiation: cell cycle arrest, modulation of the expression of cyclin-dependent kinase inhibitors, and autophagy . J Neurosurg 2003 ; 98 : 378 - 384
Yogalingam G, Pendergast AM . Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components . J Biol Chem 2008 ; 283 : 35941 - 35953
Yu SW, Baek SH, Brennan RT, Bradley CJ, Park SK, Lee YS, Jun EJ, Lookingland KJ, Kim EK, Lee H, Goudreau JL, Kim SW . Autophagic death of adult hippocampal neural stem cells following insulin withdrawal . Stem Cells 2008 ; 26 : 2602 - 2610
Yue Z, Jin S, Yang C, Levine AJ, Heintz N . Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor . Proc Natl Acad Sci USA 2003 ; 100 : 15077 - 15082
Zhou X, Ikenoue T, Chen X, Li L, Inoki K, Guan KL . Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy . Proc Natl Acad Sci USA 2009 ; 106 : 8923 - 8928
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (Mid-Career Researcher Program No. 2011-0018141).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Choi, K. Autophagy and cancer. Exp Mol Med 44, 109–120 (2012). https://doi.org/10.3858/emm.2012.44.2.033
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2012.44.2.033
Keywords
This article is cited by
-
BCL2L13 promotes mitophagy through DNM1L-mediated mitochondrial fission in glioblastoma
Cell Death & Disease (2023)
-
Polphylipoprotein-induced autophagy mechanism with high performance in photodynamic therapy
Communications Biology (2023)
-
Mechanisms of cancer cell killing by metformin: a review on different cell death pathways
Molecular and Cellular Biochemistry (2023)
-
Inhibition of autophagy by chloroquine prevents resistance to PI3K/AKT inhibitors and potentiates their antitumor effect in combination with paclitaxel in triple negative breast cancer models
Journal of Translational Medicine (2022)
-
Increase in the sensitivity to PLX4720 through inhibition of transcription factor EB-dependent autophagy in BRAF inhibitor-resistant cells
Toxicological Research (2022)
