
Abstract
C12orf65 participates in the process of mitochondrial translation and has been shown to be associated with a spectrum of phenotypes, including early onset optic atrophy, progressive encephalomyopathy, peripheral neuropathy, and spastic paraparesis.We used whole-genome homozygosity mapping as well as exome sequencing and targeted gene sequencing to identify novel C12orf65 disease-causing mutations in seven affected individuals originating from two consanguineous families. In four family members affected with childhood-onset optic atrophy accompanied by slowly progressive peripheral neuropathy and spastic paraparesis, we identified a homozygous frame shift mutation c.413_417 delAACAA, which predicts a truncated protein lacking the C-terminal portion. In the second family, we studied three affected individuals who presented with early onset optic atrophy, peripheral neuropathy, and spastic gait in addition to moderate intellectual disability. Muscle biopsy in two of the patients revealed decreased activities of the mitochondrial respiratory chain complexes I and IV. In these patients, we identified a homozygous splice mutation, g.21043 T>A (c.282+2 T>A) which leads to skipping of exon 2. Our study broadens the phenotypic spectrum of C12orf65 defects and highlights the triad of optic atrophy, axonal neuropathy and spastic paraparesis as its key clinical features. In addition, a clear genotype–phenotype correlation is anticipated in which deleterious mutations which disrupt the GGQ-containing domain in the first coding exon are expected to result in a more severe phenotype, whereas down-stream C-terminal mutations may result in a more favorable phenotype, typically lacking cognitive impairment.
Similar content being viewed by others


Introduction
The two strands of the mtDNA molecule encode 13 polypeptides, 22 tRNAs, and 2 rRNAs. The translation machinery of transcribed mtDNA consists of aminoacyl-tRNA synthetases, ribosomal protein subunits, and translation initiation, elongation, and termination factors. Although all essential mammalian mitochondrial factors involved in the initiation and elongation of translation have been cloned and well distinguished, the steps involving the termination and release process are less characterized. The mitochondrial class I release factor family is comprised of several members, including the mtRF1, mtRF1a, ICT1, and c12orf65. It was shown that in humans a single mitochondrial release factor, mtRF1a, is sufficient to release all nascent mitochondrial gene products from the mitoribosome.1 Nevertheless, the only member of the human mitochondrial release factor family implicated in human disease is c12orf65.2, 3
We report on seven new patients from two different families with confirmed novel C12orf65 mutations and further expand and delineate the clinical spectrum of C12orf65 defects.
Materials and Methods
Patient cohort
Four males and three females from two distinct consanguineous families A and B were enrolled in this study (Figure 1). The study was approved by the national and institutional review boards. All participants in this study signed a written informed consent form before enrollment (minors were signed by their parents after detailed explanations).

Pedigrees of the two families. Affected individuals are indicated by filled symbols.
Biochemical assay of OXPHOS
Enzymatic activities of the respiratory chain complexes (complexes I–IV), rotenone sensitive NADH CoQ reductase (complex I), succinate cytochrome c reductase (complex II+III), succinate dehydrogenase (complex II), cytochrome c oxidase (complex IV), and also of Mg2+ATPase (complex V) were determined in isolated muscle mitochondria from two patients (B.V-2 and B.V-3). We used the same methodology as the one used in a previous study.4 The activities were normalized to citrate synthase and compared with normal control means.
Molecular analysis
Genomic DNA was extracted from venous blood from 7 affected members and 15 unaffected members of the 2 families using the standard procedures.
Because of the well-recognized consanguinity between parents of affected individuals, a genome-wide homozygosity mapping was undertaken using the Affymetrix Gene-Chip Human Mapping 250K NspI Array (Affymetrix, Inc., Santa Clara, CA, USA) in three affected individuals from each of the two families.
We screened the entire members in the family in order to check for segregation by using specific PCR amplification and size analysis of the products using capillary electrophoresis on ABI3100 DNA sequencer (Applied Biosystem, Foster City, CA, USA) (details available upon request).
In light of the large homozygous stretch shared by the affected individuals in patients from each family and the extensive number of genes harbored in the region, we elected to perform exome sequencing in family A.
DNA sample was fragmented, end paired, adenylated, and ligated to adapters. For enrichment of exonic sequences, it was then hybridized to the SureSelect Biotinylated RNA Library, and the captured fragments were sequenced on the HiSeq 2000 platform (Illumina, San Diego, CA, USA) with mean depth coverage of 30 × . The sequenced reads were aligned, and variant calling was performed with the October 2011 release of DNAnexus software (Palo Alto,CA, USA) with the human genome assembly hg19 (GRCh37) as reference. The raw list of variants was filtered to exclude variants present in the dbSNP129, in the ‘HapMap’, and in the ‘1000 .Genomes’ databases. We specifically focused on the variants occurring in the patients’ shared homozygous region (Supplementary Material).
Sanger sequencing using customized primers was performed to validate the presence of the identified mutations in the affected patients as previously described.5
In family B, total RNA was isolated from peripheral blood lymphocytes and reverse transcribed to cDNA. The cDNA obtained was PCR-amplified using forward and reverse primers corresponding to cDNA positions c.124 and c.738 (NM_001143905.2) in exon 1 and exon 3, respectively.
The details of the specific variants described in the current paper were submitted to the Israeli National Genetic Database (http://server.goldenhelix.org/israeli/).
Results
Clinical description of patients
The main clinical features of the seven patients from our two extended consanguineous pedigrees (Figure 1) are summarized in Table 1. The most significant clinical features in our patients were optic atrophy (all patients), pes cavus (six patients), and spastic paraparesis (six patients).
Family A:
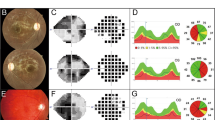
Affected subjects are three males and one female from a consanguineous family (Figure 1a). All were born after an uneventful pregnancy and delivery and had normal growth and development during their first years of life. Insidious symptoms, including visual impairment accompanied by progressive bilateral temporal optic disk pallor, were first noted at the age of 3–5 years (Figure 2a). Although bilateral optic atrophy was evident in all patients by the age of 5 years, there was significant intra-familial variability with visual acuity ranging from non-progressive mild decrease (20/40) to legal blindness (20/400). Farnsworth D-15 color vision test ranged between normal color vision and total dyschromatopsia. Visual fields were impaired, mostly with pseudo-bitemporal visual field defect in three of the four patients (Figure 2b) but with only mild or no deterioration over the years. Parents had normal ophthalmological examination, including visual acuity, color vision, and visual fields.

(a) Left eye color fundus examination of patient A.IV-1 showing temporal pallor (arrow) of the optic disc accompanied by pink (normal) nasal region. (b) Visual field study of both eyes (Humphry Treshold 30-2) in patient A.IV-1 illustrating bilateral pseudo-temporal visual field loss (arrows). (c) Peripapillary RNFL of left eye. OCT study showing generalized thinning of retinal nerve fiber more dominant on the temporal side (green color – normal thickness, yellow color – borderline thickness, red color – clear thinning). (d) Fixed flexion contracture of the toes in patient A.IV-4. Note severe atrophy of dorsal feet muscles.
Flash visual-evoked potentials were either normal or showed abnormal pattern with mild delay. Full-field electroretinogram studies were normal on photopic and scotopic stimulation. In vivo optical coherent tomography (OCT) revealed general reduction in the retinal nerve fiber layer (RNFL) thickness, mainly on the temporal part of the optic disc, consistent with the temporal disc pallor (Figure 2c). When repeated after 4 years in three of the four patients, no further reduction in RNFL thickness was observed, consistent with a very slow progression of the optic neuropathy.
By the second decade of life, affected individuals developed gait clumsiness and difficulties in regular sports activities. On examination, there was distal wasting of the calves and the feet appeared in a pes cavus position with a flexion contracture of the toes (Figure 2d), but the muscle strength remained normal. In addition, there was significant spastic paraparesis and generalized hyper-reflexia more prominent in the legs (both ankles and knees) with a bilateral plantar flexion response. In two patients (A.IV-1 and A.IV-4), mild distal sensory loss was evident in a stocking distribution in both legs. Gait abnormalities progressed very slowly allowing the three older patients independent, unaided walking at the age of 18, 20, and 23 years. Nevertheless, ankle contractures developed in two patients (A.IV-1, A.IV-4) and required daily use of ankle splints. There was no nystagmus, strabismus, ophthalmoplegia, cerebellar dysfunction, seizures, or cranial nerve deficits. All the patients graduated regular high school with no behavioral abnormalities and had normal hearing studies.
Nerve conduction velocity studies in the three older patients were consistent with severe bilateral sensory and motor peripheral neuropathy of the legs and sparing the arms. In two patients (A.IV-1, A.IV-4), lower limb action potentials were not evoked, whereas in one (A.IV-3) action potentials were severely reduced and associated with mild decrease in conduction velocity, suggesting mainly axonal damage.
Brain MRI studies done at the first and second decade of life were mostly normal in all the patients except for optic nerve atrophy, consistent with the clinical findings. Brain MR Spectroscopy (MRS) performed in patient A.IV-1 at the age of 20 years was normal and showed no abnormal lactate peak.
Family B:
One male and two females from a consanguineous family (Figure 1b) were evaluated. Following an uneventful pregnancy and labor, horizontal nystagmus was the first symptom observed in patient B.V-2 in the first months of life. Developmental milestones were globally delayed towards the end of the first year in all three patients. Although independent walking was achieved between 18 and 24 months of age, their gait was unsteady with both ataxia and spastic scissoring. In addition, hand tremor was noted on fine motor tasks in patient B.V-2. Esotropia either with or without nystagmus was initially recognized between the age of 1–4 years in all three patients, prompting either surgical repair or corrective glasses. Ophthalmological examination at that time documented progressive bilateral optic nerve pallor particularly in the temporal optic disc parts.
Disease progression was marked by a variable degree of intellectual disability ranging between mild-to-moderate intellectual disability with attention deficit and accompanied by severe optic atrophy resulting in legal blindness already during the first decade of life. In addition, all the patients developed progressive spastic paraparesis with severely limited walking accompanied by gradual foot deformities resulting in pes cavus suggesting underlying peripheral neuropathy. All three patients are already using splints to prevent further shortening of tendons while one has required surgical correction of feet deformities.
On the last follow-up at the age of 17, 12, and 8 years, all three patients had esotropia, pes cavus, lower limb hyper-reflexia, bilateral plantar flexion response, and wide-based, unsteady gait. Additionally, they had variable degree of kyphoscoliosis.
Brain MRI was normal in one patient (B.V-1) at the age of 3 years and abnormal in the other two siblings showing a hypoplastic corpus calosum with absent rostrum and splenium (Figures 3a–c) and several small and tiny hyper-intense signals on T2 and FLAIR located in the pons, in both thalami nuclei, the anterior border of the fourth ventricle, the cortico-spinal tracts, above the third ventricle, and near the tectum (Figure 3d). These lesions did not enhance by gadolinium. In one patient, two consecutive MRI studies performed 18 months apart confirmed the evolution of new lesions.

On T2 (FSE) sagittal view, (a) a short, hypoplastic corpus callosum is shown. The Rostrum is missing, and a posterior callosal hypogenesis is noted with an absence of the splenium. The optic chiasm is thin both in the sagittal (arrow head) as well as in the coronal (b) view indicating optic atrophy. Note the absence of the splenium on the T2WI axial view (c). There are bilateral high intensity foci (arrows) in the mesencephalon and the Thalamus, shown on T2 FLAIR image (d) with restricted diffusion, typical for mitochondrial disorders.
Laboratory investigations included complete blood count, routine chemistry, glucose, ammonia, thyroid functions, creatine kinase, acylcarnitines, amino acids, very long chain fatty acids, and transferrin isoelectric focusing, urinary organic acids, all of which were normal in the entire group of patients. Serum lactate was normal in all patients from family A, but showed mild-to-moderate increase (3.0–4.0 mmol/l, normal range <2.0 mmol/l) in patients from family B.
Biochemical assay of muscle OXPHOS
Muscle biopsy performed in two patients from family B (B.V-2- and B.V-3) revealed normal histology by light and electron microscopy. The enzymatic activities of the mtDNA-dependent respiratory chain complexes in isolated mitochondria from muscle tissue were partially affected; complex IV was reduced to 35 and 43% while complex I was mildly decreased to 57 and 70% in patient B.V-2 and patient B.V-3, respectively.
Molecular analysis
Based on the known consanguinity in these two families, we looked in each family for homozygous regions shared by the affected patients, yet not by their healthy siblings, using SNP arrays. We identified a single region on chromosome 12 spanning from 117 989 470 to 127 688 342 Mb and from 119 467 188 to 127 757 133 Mb (GRCh37/hg19) in families A and B, respectively (Figure 1, Supplementary Material).
In family A, the 9.7-Mb region on chromosome 12 encompassed >100 protein-coding genes, and we elected to proceed with whole-exome sequencing in patient A.IV-2. We identified 136 homozygous variants in our target chromosomal region on chromosome 12. Of these, 47 synonymous and 41 non-coding 5′ and 3′ variants were filtered out. Of the remaining 48 changes, 46 were previously annotated in dbSNP131, 1000 genome, and in the ‘HapMap’. We were left with two Sanger-validated homozygous variants: a missense mutation c.109C>T (p.(H37Y)) in VSIG10 reported in the Exome Variant Server (http://evs.gs.washington.edu/EVS/) at a frequency of 0.01% and a single homozygous deletion of five nucleotides in the C12ORF65 gene (Figure 1, Supplementary Material). The latter deletion at genomic position chr12:123741488 leads to a frame-shift mutation (c.413_417 delAACAA) (Figure 4a, left panel) and is expected to cause a premature stop codon and to result in a shortened protein of 153 amino acids instead of the wild-type 166 amino-acid protein (Figure 4c). This mutation co-segregated with the disease phenotype in all affected members of family A.

(a) Sequence analysis of C12orf65 in genomic DNA depicting a homozygous ‘AACAA’ deletion in family A (left lower panel) and a g.21043T>A (c.282+2T>A) splice mutation in family B (right upper panel). (b) Left panel shows RT-PCR using cDNA using primers corresponding to positions c.124 and c.738 (NM_001143905.2) in exon 1 and exon 3, respectively. The wild-type expected 615-bp product is shown in the third lane from the right (labeled WT). Both parents harbor the wild-type product in addition to a shorter 305-bp product (sixth and seventh lanes from the right). Both affected individuals are homozygotes for the 305-bp product (fourth and fifth lanes from the right). In addition to the major wild-type and the mutant transcripts, a 646-bp product representing an alternative transcript (NM_001194995.1) is shown in both the mother and the affected patient (sixth and fourth lanes from the right, respectively). In patient B.V-3, residual 615-bp product is evident indicating the leaky nature of the splice mutation in family B (lane 4 from the right). Following sequencing of the 305-bp product, skipping of exon 2 was identified as depicted in the right side of part b. (c) A cartoon of the C12orf65 gene (upper panel) shows exons as blocks with light blue depicting non-translated regions and dark blue-translated regions. The black stars points to the c.413_417 delAACAA mutation in family A and the donor splice mutation in family B. Lower panel shows a scheme of the encoded protein. The C12orf65 mutations are depicted as black stars with their description underneath. The GGQ domain is marked in red and the amino-acid sequence resulting from the deleted 5-nt in family A is shown in orange within the dark green block on the C-terminal part of the protein.
In family B, the biochemical assay of muscle OXPHOS together with the shared homozygous region on chromosome 12 prompted us to specifically search for a gene that may have a mitochondrial function within this region. We sequenced the C12orf65 gene and identified a homozygous mutation g.21043T>A (c.282+2 T>A) (Figure 4a, right panel). RT-PCR amplification of cDNA obtained from lymphocyte-extracted RNA in patients B.V-2- and B.V-3 demonstrated a shorter product as shown in Figure 4b. In both parents, the expected 615-bp product in addition to the mutant PCR product (305-bp) was obtained (two left lanes Figure 4b). Nevertheless, some residual wild-type transcript is amplified in B.V-3 as well as an additional transcript variant (NM_001194995.1) as described for this gene both in the mother and in patient B.V-3. Sequencing of the shorter cDNA product in both patients revealed skipping of exon 2 (Figure 4b lower panel), the first C12orf65 coding exon. The mutation showed perfect segregation in all tested family B members.
Discussion
Deleterious mutations in the C12orf65 gene were described to cause a phenotype of early onset optic atrophy, ophthalmoplegia, and progressive encephalomyopathy in three patients from two separate families.2 Recently, this gene was implicated in recessive hereditary spastic paraparesis (HSP) with optic atrophy and neuropathy.3 In the current report, we further delineate and expand the clinical and molecular spectrum of C12orf65 mutations and report seven new patients from two distinct consanguineous families harboring two novel mutations: a 5-nt deletion causing frameshift and premature truncation at position 154 instead of a full-length 166 amino-acid protein and a splice mutation affecting donor site leading to skipping of exon 2, the first coding exon (Figures 4b and c).
Taken together, the main clinical features comprising a spectrum of C12orf65 defects corresponds to a triad of optic atrophy, peripheral neuropathy, and spastic paraparesis. Accordingly, C12orf65-related defects may be classified into three phenotypes distinguished by disease severity, mortality, and the lack or presence of associated intellectual disability (Table 2).
The milder form is represented by the four subjects from family A in the current report and by the patients recently published by Shimazaki et al.3 This group displays a clinical phenotype dominated by ophthalmological impairment with slowly progressive optic neuropathy beginning within the first decade. Motor impairment due to both peripheral neuropathy and spasticity typically evolved within the second decade resulting in mild gait disturbance but, as a rule, preserved independent walking even throughout the fifth decade.3 Life expectancy is not anticipated to be shortened. Notably, none of these patients had cognitive impairment, and the brain MRI and MRS are typically normal except for optic atrophy. An intermediate phenotype is outlined by the three affected members from family B in the current report who display, in addition to early onset optic neuropathy and esotropia, variable degree of intellectual disability and progressive motor impairment dominated by both spastic and neuropathic features. Moreover, brain MRI is abnormal within the first decade, demonstrating thinning of the corpus callosum as well as gradually evolving T2 hyper-intense lesions known to be associated with mitochondrial diseases. Although too early to conclude, life expectancy does not appear to be significantly reduced. A severe phenotype with dramatically deteriorating course was described by Antonicka et al,2 dominated by significant cognitive and motor impairment resulting in wheelchair dependence within the first decade and premature death. In addition, MRI was typically abnormal with widely distributed abnormal signals. In this form, life expectancy is typically shortened resulting in premature death within the first two decades of life.
Of note, enzyme assessment of mitochondrial OXPHOS complexes both in muscle and fibroblasts typically display reduced activities of complex I and complex IV regardless of phenotype severity.
The exact role of C12orf65 in mitochondrial DNA translation is yet to be determined. C12orf65 is not directly participating in the translation machinery but is an auxiliary factor and was hypothesized to be involved in recycling of abortive peptidyl-tRNA in the mitoribosome.6
The entire group of the organellar release-factor family is characterized by the presence of a conserved peptidyl-hydrolase domain containing the universally conserved Gly-Gly-Gln (GGQ) motif,7 which interacts with the large ribosomal subunit to release the polypeptide chain from the P-site bound peptidyl-tRNA. This GGQ-containing domain stretching from amino acid 57 to 121 in the human 166-residue full-length protein8 is not affected by the mutations that lead to a relatively mild phenotype. However, both in family B and in the previously described patients with the severe phenotype2 the mutation interrupts this highly conserved domain (Figure 4c). Possible explanations for the differences between the intermediate phenotype described for the patients in family B as compared with the severe phenotype in Antonicka et al2 may be related to the leakiness nature of the novel splice mutation, which retains some residual wild-type transcript with intact exon 2 (Figure 4b – lane 4) resulting in functional protein containing the GGQ domain.
C12orf65 and ICT1, in contrast to other class I release factors, share C-terminal region comprising 20 basic residues.8 Gagnon et al9 showed that in the bacterial ICT1 homolog, YaeJ, the C-terminal tail functions as a sensor to discriminate between stalled and actively translating ribosomes. The mutations leading to a mild phenotype are expected to cause, in addition to a shortened C terminus, a less basic-charged tail, which may reduce the effectiveness of the interaction with the phosphate backbone of the ribosomal RNA that forms the walls of the ribosomal channel.
In summary, our study delineates the broad spectrum of C12orf65 defects and establishes a distinct genotype–phenotype correlation. The obligatory clinical triad is optic atrophy, peripheral neuropathy, and spastic paraparesis. Intellectual disability and behavioral abnormalities may aggravate the severity and prognosis of the disease, but its occurrence seems to be dependent on the specific region of the mutation and its severity. As the disease is progressive and symptoms may appear later in the course, we suggest to include genetic analysis of C12orf65 in the diagnostic work-up of all patients with at least two of these three major features.
References
Lightowlers RN, Chrzanowska-Lightowlers ZM : Terminating human mitochondrial protein synthesis: a shift in our thinking. RNA Biol 2010; 7: 282–286.
Antonicka H, Ostergaard E, Sasarman F et al: Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet 2010; 87: 115–122.
Shimazaki H, Takiyama Y, Ishiura H et al: A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55). J Med Genet 2012; 49: 777–784.
Saada A, Bar-Meir M, Belaiche C, Miller C, Elpeleg O : Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal Biochem 2004; 335: 66–72.
Klebe S, Lossos A, Azzedine H et al: KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet 2012; 20: 645–649.
Duarte I, Nabuurs SB, Magno R, Huynen M : Evolution and diversificationof the organellar release factor family. Mol Biol Evol 2012; 29: 3497–3512.
Seit-Nebi A, Frolova L, Justesen J, Kisselev L : Class-1 translation termination factors: invariant GGQ minidomain is essential for release activity and ribosome binding but not for stop codon recognition. Nucleic Acids Res 2001; 29: 3982–3987.
Kogure H, Hikawa Y, Hagihara M et al: Solution structure and siRNA-mediated knockdown analysis of the mitochondrial disease-related protein C12orf65. Proteins 2012; 80: 2629–2642.
Gagnon MG, Seetharaman SV, Bulkley D, Steitz TA : Structural basis for the rescue of stalled ribosomes: structure of YaeJ bound to the ribosome. Science 2012; 335: 1370–1372.
Acknowledgements
We thank the patients and their families for their participation in this study. Dr Z Argov is acknowledged for his advice and guidance. We also thank Mr Pengfei Han from Beijing Genome Institute-Shenzhen for his kind administrative assistance and Ms Batel Benporat-Zimerman and Ms Moria Gamliel for their excellent technical help. This study was sponsored in part by the Israeli MOH grant (#5914) and the Israeli MOH/ERA-Net (#4800).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Spiegel, R., Mandel, H., Saada, A. et al. Delineation of C12orf65-related phenotypes: a genotype–phenotype relationship. Eur J Hum Genet 22, 1019–1025 (2014). https://doi.org/10.1038/ejhg.2013.284
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.284
Keywords
This article is cited by
-
Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks
The Cerebellum (2017)
-
A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency
European Journal of Human Genetics (2015)
-
Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology
Human Genetics (2015)

{kind=link}