
Abstract
Tuberous sclerosis is a serious inherited disease which poses major challenges for affected families and those caring for them. Identification of the genes causing the condition and study of their protein products has shed light on the pathogenesis of the disease and provided valuable new information about signalling pathways regulating protein synthesis and cell growth. There is now the exciting possibility of drug therapy for some of the manifestations of the disease.
Similar content being viewed by others
In brief
-
Tuberous sclerosis is a multisystem disorder exhibiting a wide range of manifestations.
-
Characteristic skin lesions include facial angiofibromas, hypopigmented macules, shagreen patches and ungual fibromas.
-
Epilepsy, mental retardation and behavioural problems are often present.
-
Tumour-like lesions (hamartomas) include cortical tubers, subependymal nodules, cardiac rhabdomyomas and renal angiomyolipomas (AMLs).
-
Subependymal giant cell astrocytomas and pulmonary lymphangiomyomatosis are serious complications.
-
Inheritance is autosomal dominant with new mutations accounting for two thirds of cases.
-
A great diversity of mutations are found in the TSC1 and TSC2 genes including contiguous gene deletions of TSC2/PKD1 causing tuberous sclerosis with severe polycystic kidney disease.
-
Hamartomas result from somatic mutation following the two hit model for tumour-suppressor genes.
-
The gene products hamartin and tuberin form a dimmer, which inhibits the mammalian target of rapamycin (mTOR) in the phosphoinositide 3-kinase (PI3K) signalling pathway.
-
Inhibitors of mTOR may be of benefit in treating some aspects of the disease such as renal AMLs and pulmonary lymphangiomyomatosis. The results of the first clinical trials are awaited.
Introduction
Tuberous sclerosis (TS) is an inherited disease characterized by tumour-like lesions called hamartomas in the brain, skin, eyes, heart, lungs and kidneys.1 Epilepsy, mental retardation and autism are often present.1 The condition shows autosomal dominant inheritance and is caused by inactivating mutations in either the TSC1 or TSC2 genes. Measures of childhood prevalence range from 1 in 6800 to 1 in 17 300 but full ascertainment is difficult to achieve. Some authors use the name tuberous sclerosis complex (TSC) to emphasize the variability of the clinical features.
Clinical overview
Multisystem involvement in TS results in a wide range of manifestations.1 The commonest presentation is with seizures in infancy or early childhood, particularly infantile spasms. Partial and generalized seizures, atonic seizures (drop attacks) and myoclonic seizures also occur with the pattern of seizures evolving through childhood. A recent population-based study estimated that around 80% of children with TS have epilepsy.2 The same study gave a prevalence of mental retardation of 44% which in two-thirds of cases was profound (IQ<21). There is a strong association between mental retardation and epilepsy so that significant learning disability is very rare in patients with no history of seizures.2, 3 Risk factors for mental retardation include onset of seizures before 12 months of age, poor epilepsy control and infantile spasms. Behaviour problems are common in TS, particularly autism, autistic spectrum disorders and attention deficit hyperactivity disorder.4 Sleep disturbance is a common problem in children.
The common brain lesions in TS are tubers in the cerebral cortex and subependymal nodules along the lateral walls of the lateral ventricles.1 Histologically, tubers are disorganized areas of cortex lacking the normal laminated architecture. Large cells resembling astrocytes but positive for both glial and neuronal markers are a conspicuous feature. Subependymal nodules comprise a mixture of vascular stroma and astrocytic-like cells, some of which are large resembling those in tubers. Cranial imaging detects abnormalities in 90–95% of patients with TS.5 Computed tomography (CT) scanning is good at identifying subependymal nodules which are often calcified, but cortical tubers are better visualized by magnetic resonance imaging (MRI), particularly using fluid attenuated inversion recovery (FLAIR) sequences. Tubers may be associated with underlying white matter abnormalities such as migration lines.5 Individuals with mental retardation tend to have more tubers than those with normal intelligence3, 5 and it has been suggested that autism spectrum disorder is particularly associated with tubers in the temporal lobes.6 Subependymal nodules usually remain dormant throughout life, but have the potential to increase in size, developing into a subependymal giant cell astrocytoma (SEGA). These lesions typically occur in the head of the caudate nucleus and present with symptoms of hydrocephalus caused by obstruction to the flow of cerebrospinal fluid through the foramen of Monro. SEGAs occur in 6–14% of TS cases with the peak incidence in later childhood and adolescence.
Several types of skin lesion can occur in TS.1 Hypopigmented macules on the trunk and limbs are usually present at birth or become apparent during infancy. They are best seen in ultraviolet light using a Wood's lamp and can take any shape but are classically pointed at one end and rounded at the other resembling an ash leaf. Similar lesions on the scalp are associated with hypopigmented hair (poliosis). By 5 years of age, most children are developing angiofibromas on the face in the form of multiple flesh coloured or red papules. These typically occur over the nose, nasolabial folds, cheeks and chin. As these lesions are not related to sebaceous glands the traditional name adenoma sebaceum is inappropriate. Fibrous plaques can develop on the forehead. Shagreen patches, which are raised brown or flesh coloured connective tissue naevi, often appear on the lower back during childhood. In adolescents and adults, ungual fibromas in the form of pink or red nodules on the finger and toe nails are a common finding. Gingival fibromas also occur. Some adults with TS develop ‘confetti-like’ hypopigmentation on the limbs.
In the eyes, retinal astrocytic hamartomas are found in around half of affected individuals.7 The classic raised multinodular ‘mulberry’ lesions are easily seen, but flat, semitransparent lesions are commoner and more difficult to identify. Retinal hamartomas seldom affect vision but are a useful diagnostic sign.
Cardiac rhabdomyomas are a common finding in newborns and young children with TS and usually asymptomatic.1 They regress with age and are seldom demonstrable by adult life. Cardiac rhabdomyomas are sometimes found by chance at routine antenatal scanning and are associated with a substantial risk of TS, particularly if multiple lesions are present.8
Angiomyolipomas (AMLs) are the most common renal manifestation of TS1 developing during later childhood and adolescence. As the name implies, they are composed of blood vessels, smooth muscle and adipose tissue. The majority of adults have multiple bilateral lesions which are usually asymptomatic but can cause life-threatening haemorrhage or occasionally impaired renal function. AMLs can occur in the liver but these are rarely of clinical significance. Renal cysts are also a common finding.1 They are usually asymptomatic except in the rare case of patients with both TS and polycystic kidney disease owing to contiguous deletions of the TSC2 and PKD1 genes who usually present with severe early onset renal cystic disease and progress to end-stage renal failure by early adult life.9 Patients with TS appear to be at increased risk of renal cell carcinoma, estimated at 1–3%.10
Pulmonary lymphangioleiomyomatosis (or lymphangiomyomatosis (LAM)) is an uncommon but potentially serious manifestation of TS, which predominantly affects premenopausal women and is very rare in men.11 Normal lung is replaced by numerous cysts and histologically there is diffuse proliferation of smooth muscle cells in the lung parenchyma.
Diagnostic approaches
The diagnosis of TS is usually based on the clinical and radiological findings12 using the criteria set out in Table 1. The emphasis should be put on the major features. If two of these are present, a definite diagnosis can be made. Most of the characteristic lesions associated with TS can occasionally occur as solitary lesions, so one major feature raises the possibility of TS but does not establish the diagnosis. For example, by no means all children with giant cell astrocytomas have TS. Caution needs to be exercised when it comes to the minor features, since these are much less specific for TS. Indeed, their exclusion from the criteria would make little practical difference and achieve a useful simplification. Renal AMLs and pulmonary LAM can occur together unrelated to TS which is why other features of TS must be present before a definite diagnosis can be made.
Molecular genetic testing is not usually required to establish the diagnosis of TS. It may be helpful in patients with a possible diagnosis, particularly children too young to have developed characteristic features such as facial angiofibromas, but there is the potential pitfall of misinterpreting a rare sequence variant as pathogenic. Molecular genetic testing is not appropriate for the exclusion of TS where this diagnosis is unlikely on clinical grounds. Until the mutation detection rate approaches 100%, a negative result in this situation is of limited value. There is also the risk that in a mildly affected individual with somatic mosaicism for TS, a negative result could lead to inappropriate exclusion of the diagnosis.13 As a general rule, thorough clinical evaluation and appropriate investigations are more helpful than molecular genetic testing in resolving diagnostic difficulties.
Molecular and genetic basis of the disease
Germline and somatic mutation
TS shows autosomal dominant inheritance with approximately two-thirds of cases resulting from new dominant mutations. There is very wide variation in expression but no well documented reports of nonpenetrance. TS is caused by inactivating mutations in either the TSC1 gene at 9q3414 or the TSC2 gene at 16p13.3.15 The possibility of further TS loci on chromosomes 11 and 12 can now been discounted.
Summary data on the TSC1 and TSC2 genes and their mutation spectrum are given in Table 2. Mutation screening is costly because of the size and structure of these genes, but if methods that can detect large TSC2 deletions are included in the screening protocol, definite pathogenic mutations can be identified in 80–85% of patients.16, 17, 18 Determining whether sequence changes are pathogenic can be difficult and not always possible since both genes show a significant level of polymorphism.
Several studies have suggested that compared to sporadic cases with TSC1 mutations, sporadic cases with TSC2 mutations tend to have a more severe phenotype with a higher frequency of mental retardation and seizures, more extensive renal involvement and more severe facial angiofibromas.16, 17, 18, 19 This would explain the finding in several studies of a more equal ratio between TSC1 and TSC2 mutations in familial as compared to sporadic cases, since more severe disease in cases with new TSC2 mutations would reduce their chances of having a family. On the other hand, some missense mutations in TSC2 have been reported to be associated with particularly mild disease.20, 21
In addition to variation in severity attributable to differences between TSC1 and TSC2 mutations, there is also striking variation in expression within families. This is no doubt in part due to the random nature of somatic mutation, but the report of an association between a high expression allele of the interferon-gamma gene and a lower frequency of renal AMLs in patients with TSC2 mutations shows that modifier genes can influence the phenotype.22
Somatic mosaicism occurs in a minority of patients with TS and tends to be associated with a milder phenotype.9, 13, 23 Mutations present at low level are difficult to detect and somatic mosaicism undoubtedly explains some, if not most, of the cases where mutations cannot be identified.13, 17 Germline (confined gonadal) mosaicism is also a well established phenomenon.24
Somatic mutation accounts for the majority of lesions that occur in TS following the Knudsen two hit paradigm for tumour-suppressor genes. Cortical tubers may be an exception and it remains possible that haploinsufficiency contributes to some aspects of the phenotype. Somatic mutation often involves deletion of the chromosomal region spanning the gene, leading to loss of heterozygosity for markers in the deleted region.25, 26 Separate lesions are presumed to result from independent somatic mutations but studies in patients who have both pulmonary LAM and renal AMLs have shown matching patterns of somatic mutation in the kidney and lung lesions, supporting the hypothesis that mutant cells in both locations have a common origin.27, 28
Proteins and pathways
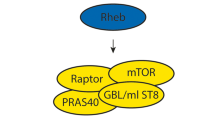
The products of the TSC1 and TSC2 genes, hamartin and tuberin, form a dimer,29, 30 which mediates a key step in the phosphoinositide 3-kinase (PI3K) signalling pathway.31 Binding of extracellular growth factors including insulin to their cell membrane receptors leads through a series of steps to phosphorylation of tuberin. This reduces the inhibitory action that the hamartin/tuberin complex exerts via the small GTPase Rheb (Ras homologue enriched in brain) on the mammalian target of rapamycin (mTOR). Activation of mTOR results in increased phosphorylation of two of its downstream targets, ribosomal S6 kinase (S6K) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1) both of which lead to increased protein translation (see legend to Figure 1 for more detail). Other signalling pathways influence the activity of the hamartin/tuberin complex which appears to have a key role in integrating information on growth stimuli, cellular energy levels, nutrient availability and hypoxia, and through mTOR regulating protein synthesis and cell growth (see Figure 1 and review by Kwiatkowski and Manning32). Functions for hamartin and tuberin independent of the complex have been suggested by evidence of their binding to a variety of other proteins, but it is uncertain whether these interactions have any physiological significance.

Role of the hamartin/tuberin complex in the mTOR signalling pathway. Extracellular growth factors binding to their cell membrane receptors lead to the recruitment of phosphoinositide 3-kinase (PI3K), which phosphorylates phosphatidyl-inositol (4,5) biphosphate (PIP2) to phosphatidyl-inositol (3,4,5) triphosphate (PIP3). This provides a membrane docking site where the protein kinase Akt is activated by phosphorylation by 3-phosphoinositide-dependent protein kinase 1 (PDK1). Akt inhibits the hamartin/tuberin complex by the phosphorylation of tuberin. The GTPase activating protein (GAP) activity of hamartin/tuberin complex inhibits Rheb-GTP by promoting conversion to Rheb-GDP. The mammalian target of rapamycin (mTOR) forms a complex with the proteins raptor (regulatory-associated protein of mTOR) and GβL. Rheb-GTP activates mTOR-raptor leading to phosphorylation of ribosomal S6 kinase (S6K) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1), increasing the activity of the former and inhibiting the latter. In both cases, the downstream effect is to promote protein synthesis and cell growth. The response to growth stimuli is modulated by other factors acting on the hamartin/tuberin complex. When energy levels are low, the presence of AMP allows LKB1 to activate AMP-activated protein kinase (AMPK) by phosphorylation, which in turn results in the phosphorylation of tuberin enhancing its GAP activity and increasing inhibition of mTOR. GAP activity is also enhanced by hypoxia acting through REDD1 and inhibited by the mitogen-activated protein kinases (MAPK) ERK1/2 and RSK1. The availability of nutrients acts on the pathway to promote protein synthesis, but it is not known whether this is by inhibition of the hamartin/tuberin complex, or by enhancement of the activity of Rheb-GTP or mTOR. In the figure, arrows denote enhanced activity and barred ends denote suppression; poorly defined pathways are indicated by a question mark and broken lines.
Mutations in TSC1 or TSC2 that impair the inhibitory function of the hamartin/tuberin complex lead to greatly increased activity of mTOR. As the name implies, mTOR is inhibited by the immunosuppressant drug rapamycin, which opens up exciting prospects for therapy. In TSC1 and TSC2 null cell lines, rapamycin restores phosphorylation downstream of mTOR to normal levels, and in rodent models of TS rapamycin and its analogues can inhibit the growth of hamartomas.33, 34 Clinical trials are now underway to investigate whether these drugs affect the progression of renal AMLs and pulmonary LAM. If so, this will have important implications for management. Other lesions that might respond to this therapeutic approach include facial angiofibromas and SEGAs. It seems unlikely that these drugs will have a role in the treatment of epilepsy or mental retardation but time will tell.
Management
Much of the management of TS is based on expert opinion since the nature of the condition makes controlled trials difficult to carry out. As a result there are significant differences in clinical guidelines and practice, particularly between countries.35, 36, 37 The commonly used evaluations and their indications are summarized in Table 3. Approaches to treatment are summarized in Table 4.
Treatment of seizures should be undertaken by a clinician experienced in the management of epilepsy. Anticonvulsants are the mainstay of therapy. The choice of medication depends on the type of seizure and cannot be covered in detail here. For infantile spasms, corticosteroids (ACTH or prednisolone) have traditionally been the first-line treatment, but in children with TS vigabatrin has been shown to be more effective.38 Unfortunately, this drug can cause constriction of the visual fields39 and the product literature advises monitoring of visual fields during treatment. Vigabatrin is not approved for use in the USA. For children with severe drug-resistant epilepsy, surgery to resect a cortical tuber acting as an epileptogenic focus can be beneficial.40 Vagal nerve stimulation may be another option, with one study suggesting some benefit.41 SEGAs can usually be treated by surgery. If fully resected the prognosis is good with a low recurrence risk.42 These tumours are rarely responsive to radiotherapy and there are reports suggesting that irradiation may induce glioblastomas. For this reason, radiotherapy is contraindicated if the lesion has been completely removed and its use in other situations needs careful consideration. There are reports suggesting benefit from regular cranial imaging to screen for SEGAs.42, 43 The expert panel at the TS Consensus Conference in 1998 concluded that early detection and treatment increased the chances of complete excision and recommended CT or MRI scanning every 1–3 years in children and less often in adults.35 This has not been universally adopted and more studies are needed to determine the value of screening and the criteria for surgical intervention.
Children diagnosed with TS need age-appropriate screening for developmental and behavioural impairments35 including autism and attention deficit hyperactivity disorder. Those affected need retesting as part of their ongoing management, particularly at preschool age to determine their educational needs and at times of transition, for example when moving school, leaving school or transferring from child to adult services. Early intervention programmes which are of benefit in children with idiopathic mental retardation and autism should help children with TS, but studies are needed to confirm this. Children with mental retardation need special educational input. Behaviour problems in TS can be difficult to manage and may need a specialist multidisciplinary approach. Stimulant medication can improve attention deficit hyperactivity disorder but may have an adverse effect on autistic behaviours and epilepsy control. Melatonin may help in children with severe sleep disorder.
Facial angiofibromas cause distress because of their appearance and are prone to bleeding. Lasers are the mainstay of treatment. The nature of the lesions determines which type of laser will give optimum results.44 This is a specialist area and treatment is best undertaken by a dermatologist experienced in the management of TS. Treatment is usually carried out in later childhood or adolescence when the lesions have stabilized but recent evidence suggests that earlier treatment may be beneficial. Ungual fibromas can be removed by excision or other means but have a tendency to recur.
Eye examination can help establish the diagnosis. Indirect ophthalmoscopy may be needed to visualize lesions in the peripheral retina. TS rarely affects vision and routine follow-up examinations are not necessary.35
Cardiac examination in infants is usually normal but sometimes identifies an arrhythmia or cardiac failure. Echocardiography is of diagnostic value in childhood but in older patients there is a risk of equivocal findings being overinterpreted. An ECG is recommended at the time of diagnosis and before surgery because this may show an abnormality before the onset of an arrhythmia.35 Treatment for cardiac rhabdomyomas is seldom required. As lesions regress, symptomatic treatment of arrhythmias or failure is the mainstay of management. Surgery is occasionally needed for intracavity lesions causing haemodynamic obstruction.
Pulmonary LAM predominantly affects adult women and no respiratory investigations are indicated in asymptomatic children or adult men.35 Chest CT is more effective than plain X-ray in detecting early LAM. It has been recommended that asymptomatic adult women with TS should be offered a one-off chest CT scan on the grounds that early diagnosis might allow symptomatic treatment to be started early.35 Hormone therapy (progesterone, tamoxifen or oophorectomy) is the mainstay of treatment.45 Lung transplantation can be life-saving for end-stage disease but recurrence in the transplanted lung has been reported.
Renal involvement can be readily assessed by ultrasound. This investigation is particularly important in newly diagnosed children to identify coexisting polycystic kidney disease.9 The majority of patients with AMLs remain asymptomatic. Intervention may be required for significant haemorrhage or suspicion of malignancy. Arterial embolization is effective in stopping haemorrhage. If surgery is required, preservation of renal tissue is an important consideration. In older children and adults ultrasound scanning at 1–3 yearly intervals has been recommended to monitor the development and progression of renal AMLs,35 particularly with a view to elective embolization of AMLs exceeding a particular size (larger than 4 cm in one study,46 exceeding 8 cm in another47). However, the indications for treatment of asymptomatic AMLs are not well established and further studies are needed to confirm the benefits of screening and validate the criteria for intervention.
Genetic counselling
Patients with TS have a 50% risk of passing on the condition to each of their offspring although the risk of a severely affected child is lower. New mutation cases are common but in advising parents it is important to confirm that neither is affected (this includes the possibility of mosaicism associated with a milder phenotype, as discussed above). If physical (including ophthalmological) examination is normal, the chances of finding evidence of TS on radiographic examination are small.48 Nevertheless it is common practice to offer renal ultrasound and cranial imaging, although the value of these investigations has been questioned.35, 49 Cranial CT may be preferable to MRI on the grounds of greater specificity and lower cost.35 For unaffected parents, the risk of another child with TS is approximately 2% because of germline mosaicism. For the siblings of new mutation cases who are clinically normal, particularly once they reach adolescence, the risk of TS is very small and it is debatable whether any investigations are needed. If the TS mutation can be identified in the index case, genetic testing can be offered to the parents and siblings to confirm their disease status.
Prenatal diagnosis
If the mutation in the index case is known, first trimester chorionic villus sampling and molecular genetic diagnosis is an option. If this is not possible, echocardiography to identify cardiac rhabdomyomas may detect an affected pregnancy but a normal echo does not exclude the diagnosis. Ultrafast MRI can detect intracranial lesions as early as 21 weeks gestation50 but studies are needed to determine the sensitivity and specificity of this approach.
Conclusion
Encouraging progress has been made over the past decade in meeting the challenges posed by this serious and multifaceted disorder. There have been many improvements in medical management but there is still little that can be offered to children with mental retardation or intractable epilepsy. More work is needed to develop evidence-based screening protocols. The genetic basis for the disease is now well understood and genetic testing is available for the majority of families, but better methods need to be developed for rapid and reliable identification of pathogenic mutations. Remarkable advances have been made in our understanding of the pathogenesis of TS. There is now the exciting prospect of drug therapy for some serious aspects of the disorder and the results of the first clinical trials are eagerly awaited.
References
Further reading
Curatolo P (ed): Tuberous Sclerosis Complex: From Basic Science to Clinical Phenotypes. London: Mac Keith Press, 2003.
Guertin DA, Sabatini DM : An expanding role for mTOR in cancer. Trends Mol Med 2005; 11: 353–361.
Inoki K, Corradetti MN, Guan KL : Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005; 37: 19–24.
Yates JRW : Tuberous Sclerosis; in Cassidy SB, Allanson JE (eds): Management of Genetic Syndromes. Hoboken, New Jersey: John Wiley & Sons, 2005.
Many of the national tuberous sclerosis patient associations produce useful leaflets and other resources for patients and professionals. Their contact details can be obtained from the Tuberous Sclerosis International website at http://www.stsn.nl/tsi/tsi.htm.
References
Gomez MR, Sampson JR, Whittemore VH (eds): Tuberous Sclerosis Complex. Oxford: Oxford University Press, 1999.
Joinson C, O'Callaghan FJ, Osborne JP, Martyn C, Harris T, Bolton PF : Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med 2003; 33: 335–344.
O'Callaghan FJ, Harris T, Joinson C et al: The relation of infantile spasms, tubers, and intelligence in tuberous sclerosis complex. Arch Dis Child 2004; 89: 530–533.
Gillberg IC, Gillberg C, Ahlsen G : Autistic behaviour and attention deficits in tuberous sclerosis: a population-based study. Dev Med Child Neurol 1994; 36: 50–56.
Shepherd CW, Houser OW, Gomez MR : MR findings in tuberous sclerosis complex and correlation with seizure development and mental impairment. Am J Neuroradiol 1995; 16: 149–155.
Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A : Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain 2002; 125: 1247–1255.
Rowley SA, O'Callaghan FJ, Osborne JP : Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol 2001; 85: 420–423.
Tworetzky W, McElhinney DB, Margossian R et al: Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol 2003; 92: 487–489.
Sampson JR, Maheshwar MM, Aspinwall R et al: Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet 1997; 61: 843–851.
Nelson CP, Sanda MG : Contemporary diagnosis and management of renal angiomyolipoma. J Urol 2002; 168: 1315–1325.
Hancock E, Tomkins S, Sampson J, Osborne J : Lymphangioleiomyomatosis and tuberous sclerosis. Respir Med 2002; 96: 7–13.
Roach ES, Gomez MR, Northrup H : Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998; 13: 624–628.
Kwiatkowska J, Wigowska-Sowinska J, Napierala D, Slomski R, Kwiatkowski DJ : Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N Engl J Med 1999; 340: 703–707.
van Slegtenhorst M, de Hoogt R, Hermans C et al: Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277: 805–808.
The European Chromosome 16 Tuberous Sclerosis Consortium: Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75: 1305–1315.
Jones AC, Shyamsundar MM, Thomas MW et al: Comprehensive mutation analysis of TSC1 and TSC2 – and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 1999; 64: 1305–1315.
Dabora SL, Jozwiak S, Franz DN et al: Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001; 68: 64–80.
Sancak O, Nellist M, Goedbloed M et al: Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype – phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet 2005; 13: 731–741.
Lewis JC, Thomas HV, Murphy KC, Sampson JR : Genotype and psychological phenotype in tuberous sclerosis. J Med Genet 2004; 41: 203–207.
Mayer K, Goedbloed M, van Zijl K, Nellist M, Rott HD : Characterisation of a novel TSC2 missense mutation in the GAP related domain associated with minimal clinical manifestations of tuberous sclerosis. J Med Genet 2004; 41: e64.
O'Connor SE, Kwiatkowski DJ, Roberts PS, Wollmann RL, Huttenlocher PR : A family with seizures and minor features of tuberous sclerosis and a novel TSC2 mutation. Neurology 2003; 61: 409–412.
Dabora SL, Roberts P, Nieto A et al: Association between a high-expressing interferon-gamma allele and a lower frequency of kidney angiomyolipomas in TSC2 patients. Am J Hum Genet 2002; 71: 750–758.
Verhoef S, Bakker L, Tempelaars AM et al: High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet 1999; 64: 1632–1637.
Yates JR, van Bakel I, Sepp T et al: Female germline mosaicism in tuberous sclerosis confirmed by molecular genetic analysis. Hum Mol Genet 1997; 6: 2265–2269.
Green AJ, Johnson PH, Yates JR : The tuberous sclerosis gene on chromosome 9q34 acts as a growth suppressor. Hum Mol Genet 1994; 3: 1833–1834.
Green AJ, Smith M, Yates JR : Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet 1994; 6: 193–196.
Carsillo T, Astrinidis A, Henske EP : Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA 2000; 97: 6085–6090.
Yu J, Astrinidis A, Henske EP : Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am J Respir Crit Care Med 2001; 164: 1537–1540.
Plank TL, Yeung RS, Henske EP : Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res 1998; 58: 4766–4770.
van Slegtenhorst M, Nellist M, Nagelkerken B et al: Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 1998; 7: 1053–1057.
Manning BD, Cantley LC : Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 2003; 28: 573–576.
Kwiatkowski DJ, Manning BD : Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 2005; 14 (Spec no. 2): R251–R258.
Kenerson H, Dundon TA, Yeung RS : Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res 2005; 57: 67–75.
Lee L, Sudentas P, Donohue B et al: Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer 2005; 42: 213–227.
Roach ES, DiMario FJ, Kandt RS, Northrup H : Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 1999; 14: 401–407.
The Tuberous Sclerosis Association: Clinical guidelines for the care of patients with tuberous sclerosis complex http://www.tuberous-sclerosis.org/professionals/guidelines.shtml.
Scottish Clinical Genetics Guidelines Working Group: Clinical guidelines for tuberous sclerosis, 1998, http://www.genisys.hw.ac.uk/cgi-bin/WebObjects/genisys.
Hancock E, Osborne JP : Vigabatrin in the treatment of infantile spasms in tuberous sclerosis: literature review. J Child Neurol 1999; 14: 71–74.
Vanhatalo S, Nousiainen I, Eriksson K et al: Visual field constriction in 91 Finnish children treated with vigabatrin. Epilepsia 2002; 43: 748–756.
Jarrar RG, Buchhalter JR, Raffel C : Long-term outcome of epilepsy surgery in patients with tuberous sclerosis. Neurology 2004; 62: 479–481.
Parain D, Penniello MJ, Berquen P, Delangre T, Billard C, Murphy JV : Vagal nerve stimulation in tuberous sclerosis complex patients. Pediatr Neurol 2001; 25: 213–216.
Torres OA, Roach ES, Delgado MR et al: Early diagnosis of subependymal giant cell astrocytoma in patients with tuberous sclerosis. J Child Neurol 1998; 13: 173–177.
Goh S, Kwiatkowski DJ, Dorer DJ, Thiele EA : Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology 2005; 65: 235–238.
Papadavid E, Markey A, Bellaney G, Walker NP : Carbon dioxide and pulsed dye laser treatment of angiofibromas in 29 patients with tuberous sclerosis. Br J Dermatol 2002; 147: 337–342.
Johnson SR, Tattersfield AE : Clinical experience of lymphangioleiomyomatosis in the UK. Thorax 2000; 55: 1052–1057.
Kothary N, Soulen MC, Clark TW et al: Renal angiomyolipoma: long-term results after arterial embolization. J Vasc Interv Radiol 2005; 16: 45–50.
Dickinson M, Ruckle H, Beaghler M, Hadley HR : Renal angiomyolipoma: optimal treatment based on size and symptoms. Clin Nephrol 1998; 49: 281–286.
Roach ES, Kerr J, Mendelsohn D, Laster DW, Raeside C : Detection of tuberous sclerosis in parents by magnetic resonance imaging. Neurology 1991; 41: 262–265.
Fryer AE, Chalmers AH, Osborne JP : The value of investigation for genetic counselling in tuberous sclerosis. J Med Genet 1990; 27: 217–223.
Levine D, Barnes P, Korf B, Edelman R : Tuberous sclerosis in the fetus: second-trimester diagnosis of subependymal tubers with ultrafast MR imaging. Am J Roentgenol 2000; 175: 1067–1069.
Acknowledgements
I thank Dr Geoff Woods for his helpful comments on the draft manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yates, J. Tuberous sclerosis. Eur J Hum Genet 14, 1065–1073 (2006). https://doi.org/10.1038/sj.ejhg.5201625
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201625
Keywords
This article is cited by
-
Clinical, genetic and quality-of-life study of a cohort of adult patients with tuberous sclerosis
Orphanet Journal of Rare Diseases (2021)
-
mTOR inhibitors in the pharmacologic management of tuberous sclerosis complex and their potential role in other rare neurodevelopmental disorders
Orphanet Journal of Rare Diseases (2017)
-
Analysis of genotypes and phenotypes in Chinese children with tuberous sclerosis complex
Science China Life Sciences (2017)
-
Treatment of renal angiomyolipoma in tuberous sclerosis complex (TSC) patients
Pediatric Nephrology (2017)
-
Neurocutaneous Disorders for the Practicing Neurologist: a Focused Review
Current Neurology and Neuroscience Reports (2016)
