
Abstract
Understanding of the movements of species at multiple scales is essential to appreciate patterns of population connectivity and in some cases, the potential for pathogen transmission. The serotine bat (Eptesicus serotinus) is a common and widely distributed species in Europe where it frequently harbours European bat lyssavirus type 1 (EBLV-1), a virus causing rabies and transmissible to humans. In the United Kingdom, it is rare, with a distribution restricted to south of the country and so far the virus has never been found there. We investigated the genetic structure and gene flow of E. serotinus across the England and continental Europe. Greater genetic structuring was found in England compared with continental Europe. Nuclear data suggest a single population on the continent, although further work with more intensive sampling is required to confirm this, while mitochondrial sequences indicate an east–west substructure. In contrast, three distinct populations were found in England using microsatellite markers, and mitochondrial diversity was very low. Evidence of nuclear admixture indicated strong male-mediated gene flow among populations. Differences in connectivity could contribute to the high viral prevalence on the continent in contrast with the United Kingdom. Although the English Channel was previously thought to restrict gene flow, our data indicate relatively frequent movement from the continent to England highlighting the potential for movement of EBLV-1 into the United Kingdom.
Similar content being viewed by others

Introduction
Bats are widely distributed and vary enormously in their ecology, sociality and behaviour. Largely because of their small size and vagility, population genetics has been widely used to infer aspects of bat biology that are difficult to observe directly. Substantial variation in genetic structuring has been found, reflecting differences in bat movement, behaviour, mating and social systems (Burland and Wilmer, 2001; Moussy et al., 2013). In addition, molecular approaches have been used to assess many aspects of bat ecology including potential sex-biased dispersal and movements (Petit et al., 2001), mating systems (Furmankiewicz and Altringham, 2007), effect of landscape barriers (García-Mudarra et al., 2009), demography and history (Flanders et al., 2009) and sociality (Metheny et al., 2007). Gaining an understanding of these components of bat ecology and behaviour is fundamental in a conservation context (Racey and Entwistle, 2003), to improve our understanding of ecosystem services (Kunz et al., 2011) and in disease epidemiology because bats are now thought to be the wildlife reservoirs for a range of important zoonoses and emerging diseases (Kuzmin et al., 2011).
The serotine bat, Eptesicus serotinus, is a common species within its core range in continental Europe, but rare in the United Kingdom where it is largely restricted to southern England (Dietz et al., 2009). The species is considered sedentary in the United Kingdom with recorded seasonal flights of under 100 km, although ringing data are sparse, which restricts our understanding of any seasonal movements (Hutterer et al., 2005). Adult females form maternity colonies in the summer, often in inhabited buildings, and display some level of philopatry to their natal landscape, if not natal colony (Harbusch, 2003). Owing to the relative ease of locating these maternity roosts, research has mainly focused on habitat and roost use, diet, and activity patterns of adult females or of juveniles of both sexes (Catto et al., 1996; Robinson and Stebbings, 1997). In contrast, adult males are more difficult to locate and their ecology is very poorly understood. Similarly, little is known about the winter ecology of the species and its mating system. Information on serotine social and spatial organisation of as well as on population and seasonal connectivity is required for a more complete understanding of its ecology and behaviour within its core range and at the edge of its distribution.
This basic information on serotine biology is particularly important as this bat is the main reservoir for the most common European bat lyssavirus, EBLV-1 (Harris et al., 2006). In such a context, knowledge of the host’s movement, mating patterns and social organisation is crucial to gain further understanding of viral dynamics. Currently, the prevalence of EBLV-1 in continental Europe contrasts strongly with its apparent absence from England (Schatz et al. 2013). Indeed, both active and passive surveillance in the United Kingdom has failed to detect the virus. However, passive surveillance is biased to common species with only few E. serotinus submitted for testing. Furthermore, a juvenile bat was tested positive for antibodies against EBLV-1 in a maternity roost of south of England (Harris et al. 2009). This case was surprising as the bat had therefore not dispersed or mated, and was therefore likely to have only been in contact with roost-mates, but no other individuals from the same roost was seropositive despite extensive sampling over 4 years.
Here, we used 10 microsatellites and a portion of the hypervariable region II of mitochondrial DNA (mtDNA) to investigate the genetic structure of E. serotinus at multiple spatial scales. We initially describe in detail the genetic structure of serotines in England and provide a first account of the level of genetic differentiation among locations on the continent to compare the core and edge populations. Based on the bat’s biology, two contrasting predictions were made. First, because the English population is small and fragmented at the edge of the species range (Bat Conservation Trust, 2013), movement and gene flow could be reduced there, resulting in genetic differentiation and structure. If the core continental population were also large and continuous enough to result in low structure, then these contrasting structure patterns could help explain the prevalence of EBLV-1 on the continent and its apparent absence in England—bats simply do not move enough in the United Kingdom to spread the virus. Alternatively, because the geographic area covered by the English population is small and there are few obvious barriers to movement within the United Kingdom, it could display low structure and low genetic differentiation. If this were so, then the apparent absence of EBLV-1 in the United Kingdom could simply be due limited dispersal across the English Channel. We also tested for possible sex-biased gene flow as male connectivity and female philopatry are common in temperate bats (Moussy et al., 2013).
Materials and methods
Sample collection
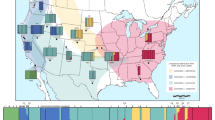
A total of 593 individuals were captured at 28 maternity roosts in inhabited buildings across the English range in the summers of 2004–2006 and 2010–2011 (individuals per roosts: mean=19.41; median=14.50; range from 4 to 68; Supplementary Table S1; Figure 1). No adult males were captured because of their absence from maternity roosts. A wing tissue sample was taken using a 3 mm biopsy punch (Stiefel Laboratories, Wooburn Green, UK) and stored in 70% ethanol at +4 °C until DNA extraction. All bats were released back into the wild within a few minutes of the procedure at their location of capture. All work was done with the approval of ethical review bodies at FERA and University of Exeter and under licence from the UK Home Office (Animals (Scientific Procedures) Act 1986) and Natural England (Habitats Regulations, 2010). Wing biopsies were also supplied for 106 individuals from eight locations in six countries across continental Europe (Supplementary Table S1; Figure 1).

Geographical location of the Eptesicus serotinus roosts sampled in Europe (inset) and in the UK. The shading represents the known species distribution (Dietz et al., 2009). CH, Switzerland; FR, France; GG, Georgia; IT, Italy; PL, Poland; SL, Slovakia; UK, England.
DNA extraction and whole-genome amplification
DNA was extracted following Sambrook and Russel (2001). Ammonium acetate (10 M; Sigma-Aldrich, Poole, UK) was used to precipitate proteins instead of potassium acetate. The DNA pellet was recovered in 1 × Tris-ethylenediaminetetraacetic acid buffer (TE) (Sigma-Aldrich) and stored at −20 °C before further procedures. The whole-genome of each individual was amplified using illustra GenomiPhi V2 DNA Amplification kit (GE Healthcare, Little Chalfont, UK). A subset of samples was genotyped for microsatellites before and after whole-genome amplification to ensure the procedure did not affect typing success or quality.
Microsatellite genotyping and mtDNA sequencing
Samples were genotyped using a panel of 11 microsatellite markers (Smith et al., 2011) as described in Supplementary Table S2. PCR was carried out in 15 μl, with 7.5 μl 2x PCR mastermix (ABgene, Epsom, UK) containing 1.5 mM MgCl2, 1 μM of each primer (Sigma-Aldrich/Applied Biosystems, Foster City, CA, USA), and 1.5 μl DNA template (DNA extract or 1/10 dilution in 1x TE of whole-genome amplification product). The remaining volume was made up with molecular grade water. PCR reactions consisted of denaturation at 94 °C for 2 min, followed by 35 cycles of 94 °C for 1 min, 1 min at the annealing temperature (Supplementary Table S2), 72 °C for 1 min, and final elongation at 72 °C for 5 min and 60 °C for 1 h. PCR products were diluted and mixed into four sets (Supplementary Table S2) and run on an ABI Prism 3130xl genetic analyser (Applied Biosystems) with Genescan Rox 500 size standard (Applied Biosystems). Microsatellite alleles were sized using GeneMapper 3.7 software (Applied Biosystems). The genotyping error rate per loci was estimated by re-amplifying and re-genotyping 11–24% of samples depending on the locus.
A 460-bp portion of the hypervariable domain II of the mtDNA control region was amplified in a subset of samples (Supplementary Table S1) using primers for conserved sequence block F on L-strand 5′-CTACCTCCGTGAAACCAGCAAC-3′ (Wilkinson and Chapman, 1991) and for hypervariable domain II on H-strand 5′-CGTACACGTATTCGTATGTATGTCCT-3′ (J Juste, personal communication). PCR was carried out in 20 μl, with 10 μl 2x BioMix Red (Bioline, London, UK) containing 1 mM MgCl2, 0.5 μM of each primer (Sigma-Aldrich), and 1 μl of amplified genomic DNA. The remaining volume was made up with molecular grade water. PCR reactions consisted of denaturation at 94 °C for 5 min, followed by 30 cycles of 94 °C for 1 min, 55 °C for 1.5 min, 72 °C for 2 min, and final elongation at 72 °C for 5 min and 60 °C for 1 h. PCR products were purified by an enzymatic reaction to remove leftover primers and dNTPs. A 10 μl mix of 0.5 U Exonuclease I (Fermentas, Vilnius, Lithuania) and 0.25 U alkaline phosphatase (Fermentas) in molecular grade water was added to each product before incubation at 37 °C for 30 min and deactivation at 85 °C for 15 min. The purified DNA fragment was sequenced from both directions using the BigDye Terminator v3.1 sequencing kit (Applied Biosystems) in a 20 μl reaction volume with 1.5 μl BigDye Terminator mix, 3.5 μl 5x sequencing buffer, 3.33 pmol of primer and 1 μl DNA template. The remaining volume was made up with molecular grade water. The sequencing reaction consisted of 25 cycles of 96 °C for 10 s, 50 °C for 5 s and 60 °C for 4 min. The sequencing reactions were cleaned by precipitation with 100% ethanol and 20 μg glycogen before a washing step with 70% ethanol. They were then reconstituted in 11 μl HiDi formamide before being run on an ABI Prism 3130xl genetic analyser (Applied Biosystems). The resulting sequences were assembled, aligned and trimmed into GENEIOUS 5.5.7 (Drummond et al., 2011) to create a 424-bp consensus sequence for each individual.
Microsatellites analysis: tests of assumptions, genetic diversity and basic statistics
The presence of scoring inconsistencies, null alleles, large allele dropout and stuttering were tested in MICROCHECKER (Van Oosterhout et al., 2004). Tests for departure from Hardy–Weinberg equilibrium were performed for each roost and each locus in GENEPOP 4.1.4 (Rousset, 2008) both as two-tailed tests with no assumption as the direction of the deviation, and under the hypothesis of heterozygote deficit. A sequential Bonferonni correction was applied to the P-values (Rice, 1989).Tests of linkage disequilibrium were performed in FSTAT 2.9.3.2 (Goudet, 1995, 2001) and the significance levels were adjusted using sequential Bonferonni correction. Genetic diversity indices were obtained for each roost from FSTAT and GENETIX 4.05 (Belkhir et al., 1996). Estimated FST averaged over loci (Weir and Cockerham, 1984) and the 95% confidence intervals (CIs) after 1000 bootstrap over loci were calculated in GENETIX. Finally, a log-likelihood G-test for population differentiation (not assuming random mating within roosts) was performed (Goudet et al., 1996) in FSTAT based on 100 000 randomisations of complete multilocus genotypes.
Nuclear population structure
The Bayesian clustering program STRUCTURE was used to investigate genetic structure (Pritchard et al., 2000). We evaluated the number of assumed clusters K from 1 to 6 using a burn-in of 5 × 106 iterations followed by 5 × 107 Markov Chain Monte Carlo iterations. The admixture model with correlated allele frequencies was run with and without population information (roosts the individuals were sampled from) as a prior. Each K value was run 12 times to ensure stability and convergence of the chains. The optimal number of clusters K was obtained from ΔK, based on the rate of change in the log probability of data in successive K values (Evanno et al., 2005) as implemented on STRUCTURE HARVESTER (Earl and VonHoldt, 2012).
Genetic structuring was further investigated using two multivariate methods: (1) a discriminant analysis of principal components (Jombart et al., 2010) and (2) a spatial principal component analysis (sPCA) (Jombart et al., 2008). In contrast to Bayesian clustering, the strength of these approaches rests in their independence from population genetic model, and inferences are therefore made only on allelic similarity (Jombart et al., 2009). The two methods were implemented in the ADEGENET 1.3-4 package (Jombart, 2008) in R (R Core Team, 2014). Discriminant analysis of principal component has been developed to summarise the overall genetic variability of individuals within groups while optimising discrimination between groups. We used the roosts individuals were sampled from as group before discriminant analysis. The first 30 principal components (PC) of PCA were retained in the data transformation step, covering 90.5% of genetic variance. sPCA explicitly incorporates spatial information in the investigation of genetic variability across landscapes (Jombart et al., 2008). We applied this method at the roost level. A connection network was constructed using K-nearest neighbour (Cover and Hart, 1967), setting K to seven to reflect the number of sites sampled on the continent. This network was used for the calculation of Moran’s I (Moran, 1948, 1950). sPCA optimises the product of the variance of individual scores, based on roosts’ allelic frequencies, and of Moran’s I, in order to summarise genetic variability in a spatial context.
AMOVA and genetic differentiation among clusters
Consensus on the population structure was reached based on results from the Bayesian and multivariate analyses. Roosts were assigned to one of the clusters defined (hereafter called populations) and this genetic structure was tested in an analysis of molecular variance (AMOVA) (Excoffier et al., 1992) in ARLEQUIN (Excoffier and Lischer, 2010). Log-likelihood G tests for differentiation were performed between and within populations using 10 000 permutations in the HIERFSTAT package implemented in R. Pairwise FST between the populations were obtained from ARLEQUIN to assess the level of connectivity. Log-likelihood G tests were performed for all pairs of populations on 10 000 permutations by the HIERFSTAT package.
Isolation by distance
Mantel and partial Mantel tests were performed between matrices of linearised pairwise FST (Rousset, 1997) and log-transformed geographical distances among roosts. An indicator matrix was included to account for the presence of the most obvious geographical barriers including the English Channel (isolating the United Kingdom), the Solent (between the Isle of Wight (IOW) and England), the Alps (isolating the Italian roost), the Carpathians (isolating the Slovakian roost) and the Caucasus range (isolating Georgia). Tests were carried out on the full data set, the continent only data set and on the English-only data set.
Contemporary gene flow
The magnitude and direction of contemporary of gene flow occurring between the consensus populations were estimated using the program BAYESASS 3.0.1 (Wilson and Rannala, 2003). The simulation was run with 107 iterations, with the first 25 × 105 iterations discarded as burn-in. Samples were collected every 100 iterations. Allelic frequencies and inbreeding coefficients were set at 0.2, whereas migration rate was left at its default value of 0.1. These mixing parameters ensured the acceptance rates were between 0.2 and 0.4. The trace file was examined in TRACER 1.5 to ensure mixing and convergence of the chains (Rambaut and Drummond, 2009).
MtDNA analysis: genetic diversity and differentiation
Haplotype (h) and nucleotide diversities (π), number of polymorphic sites and mean number of pairwise differences were calculated for each roost and each population in ARLEQUIN. Differentiation index for haploid data ϕST (Weir and Cockerham, 1984) was estimated in ARLEQUIN and tested for significant departure from panmixia after 10 000 permutations. Differentiation test among roosts was also performed with 100 000 MC iterations after 10 000 burn-in iterations in ARLEQUIN.
Mitochondrial population structure
Spatial structuring of genetic variation in the mtDNA sequences was investigated by sPCA (Jombart et al., 2008). All roosts were considered as neighbours in this analysis, but their interconnection was weighted by the inverse Euclidian distance separating the roosts.
Geographical structuring of mtDNA variation was also investigated in an AMOVA. Roosts were grouped in various clusters based on the sPCA results and on the structure observed at the nuclear level. Those structures were tested in order to optimise the among groups variance component. The contribution of each hierarchical component (among groups, among roosts within groups and within roosts) was estimated and tested for significance after 10 000 permutations. Differentiation between identified clusters was inferred by pairwise ϕST and tested by log-likelihood tests with 100 000 MC iterations after 10 000 burn-in iterations. All calculations were undertaken in ARLEQUIN.
Mantel and partial Mantel tests were performed between matrices of pairwise linearised ϕST and log-transformed geographical distances among roosts to test for isolation by distance. The same indicator matrix used to infer presence of geographical barriers in the microsatellite data set was included. Tests were carried out on the full data set, the continent only data set and on the English data set.
Phylogenetic analysis
Phylogenetic reconstructions were undertaken on unique haplotypes by maximum-likelihood (ML) criteria (Beerli and Felsenstein, 2001) and a Bayesian approach. The K80+I model (Kimura, 1980) of DNA substitution (transition/transversion ratio=18.374, proportion of invariable sites=0.817) was applied to the ML and the Bayesian analyses as determined by the program jMODELTEST 0.1.1 (Wu and Eisen, 2008; Posada, 2008) using Bayesian Information Criterion. The ML analysis was undertaken in PHYML 3.0 (Wu and Eisen, 2008) and the Bayesian inference in MRBAYES 3.1 (Ronquist and Huelsenbeck, 2003). For the Bayesian analysis, four chains of 107 iterations each were run, taking samples every 100 generations and the first 25% of trees were discarded as burn-in. The statistical support of the branching pattern obtained by ML approach was assessed by 1000 bootstrap replicates. In addition, a haplotype network (Excoffier and Smouse, 1994) was constructed using ARLEQUIN and visualised in HAPSTAR (Teacher and Griffiths, 2011). Demographic history was inferred from Fu’s FS, a measure of marker neutrality sensitive to changes in population sizes, and mismatch distributions. Populations that have undergone recent expansion display significantly negative FS value (Fu and Li, 1993) and a smooth or unimodal distribution of pairwise differences among haplotypes (Schneider and Excoffier, 1999). These analyses were run in ARLEQUIN with 10 000 bootstrap replicates to generate the simulated mismatch distribution in a population expansion model and 10 000 randomised samples to test for FS significance.
Results
Microsatellites
Tests of assumption, genetic diversity and basic statistics
Of the 11 microsatellites genotyped, one (AF141650) demonstrated high levels of estimated null alleles, genotyping inconsistencies and significant deviation from the assumption of Hardy–Weinberg equilibrium in a large number of sampled roosts. It was thus removed from subsequent analyses. Of the remaining loci, three deviated from Hardy–Weinberg equilibrium in 1 to 4 roosts out of 36 and 6 showed low-to-moderate levels of estimated null alleles. Deviation from Hardy–Weinberg expectations was due to heterozygote deficiency and as it was noticed in only a few roosts, it could be caused by factors other than by selection on the markers. No significant linkage disequilibrium between loci was detected after sequential Bonferonni correction. The 10 remaining microsatellites were thus used for subsequent analyses. The mean genotyping error rate for the 10 markers was 4.21% (±2.50%). Mean observed and expected heterozygosity and within roost gene diversity were moderate and consistent across all roosts (Ho=0.540±0.06; HE=0.572±0.04; HS=0.604±0.04; Supplementary Table S3), and allelic richness was low but still similar across all roosts (AR=2.9±0.1; Supplementary Table S3). The continent had significantly higher allelic richness than England (P<0.001), but none of the other genetic diversity measures differed significantly between the two (HO: P=0.594; HE: P=0.196, HS: P=0.127, FIS: P=0.721)
Estimated FST averaged over loci was low at 0.048 but significant (0.039–0.059 95% CI). Roosts were found to be significantly genetically differentiated (randomisation test: P<0.001), which in combination with the significant structure, suggests the roosts sampled were not part of a single panmictic population.
Bayesian modelling
Despite high levels of admixture, clustering was consistent with the sampling geography. At K=2, the sites on the continent and eastern England formed one group and those in western England and IOW another. At K=3, the continent was discriminated from eastern England. At K=4 and 5, the IOW was distinct (Figure 2 and Supplementary Figure S1). The standardised second-order rate of change ΔK indicated that the most likely number of clusters describing the data were three (Supplementary Figure S1). As this method only distinguished higher structuring level, each cluster was then run separately with the same settings. No obvious geographical substructure was supported for the continent or for eastern England. In contrast, substructure was revealed in the third cluster, with roosts on the IOW segregating from western England.

Individual-based analyses on microsatellite data using multivariate (a and b) and Bayesian (c) assignment methods. (a) Scatterplot of individual principal components on the first two axes. Each roost is colour coded. (b) Posterior probability of membership to one of the roosts after discriminant analysis on 30 PCs using roosts as a prior and leave-one out cross-validation. Each vertical line represents an individual. Solid lines separate roosts the individuals were sampled from. The roosts are ordered according to their geographical location and colour coded as in a. (c) Bayesian posterior probability of membership to one of four populations obtained by STRUCTURE, using sampling roosts as prior and admixture settings. Individuals and roosts are displayed in the same order as in b.
Multivariate analysis
The discrimination between roosts using discriminant analysis of PCs was low, reflecting high within roost genetic variability and high admixture levels (91.72% of individuals have membership probabilities to the sampling roost <90%). Despite this admixture, the first two PCs captured 42% of the genetic structure. PC-1 mostly delineated roosts from the continent and the IOW from those on mainland England, whereas PC-2 discriminated roosts from west of England and the IOW from those on the continent and in east of England, a pattern of clustering similar to the STRUCTURE analysis (Figure 2).
Population structure was also inferred by sPCA. The three first axes had the highest eigenvalues and were therefore retained. PC-1 mostly segregated England from the continent. PC-2 differentiated eastern England from western England and western continental sites from Eastern Europe. The continental sPCA scores were low compared with English ones, suggesting that the continental structuring was weak compared with that across England. PC-3 retrieved a final structure by clustering sites in France with the IOW and Central England (Supplementary Figure S2). Thus, the United Kingdom was clearly differentiated from the continent and subdivided into three groups representing the east, the west and the IOW (Figure 3).

Scores from the spatial principal component analysis (sPCA) on microsatellites in Europe (a) and in England (b) and on mtDNA sequence in Europe (c) and in England (d). sPCA optimised the product of the roost genetic variances and of Moran's I for the two (mtDNA) or three (microsatellites) axes retained. A neighbouring network was built as a prior using K nearest neighbour=7 for microsatellites and the inverse geographical distance for mtDNA. Each roost was mapped by colour coding its two or three sPCA lagged scores as intensity of a given colour channel (first axis: red, second axis: green, third axis: blue).
AMOVA and genetic differentiation among clusters
Four populations were retained through consensus from the previous analyses—continental Europe, eastern England, western England and the IOW (Supplementary Table S2). Differentiation tests were significant both between and within populations (FCT=0.034, P=0.0001; FSC=0.025, P=0.0001), suggesting both the stratification of the genetic variation and the importance of roosts as structuring units within those clusters. Pairwise FST suggested a closer relationship between the continent and eastern England than with the other clusters, a very close relationship between eastern and western England, and a stronger connection between the IOW and western England than to the other clusters. The IOW was also more connected to the continent than to eastern England (Table 1). Differentiation was significant for all pairwise connections with the exception of east–west England (P=0.510) and continent-IOW (P=0.652).
Isolation by distance
Genetic and geographical distances were significantly correlated when controlling for the effects of physical barriers on the full data set (r=0.214, P=0.021), on the continental data set (r=0.463, P=0.024) and on the English data set (r=0.302, P<0.001) (Supplementary Figure S3). The physical barriers tested also had a significant effect on genetic differentiation when controlling for geographical distance at all scales (Europe: r=0.373, P<0.001; Continent: r=0.511, P=0.024; England: r=0.560, P<0.001).
Contemporary gene flow
Roosts were pooled into the four populations retained from previous analyses, and recent migration rates were estimated as the mean proportion of individuals moving between populations (Table 2). The continental population displayed the highest emigration rate and the lowest immigration rate, indicating that contemporary gene flow over the Channel is mostly biased toward England. Interestingly, the IOW seems to have an important role in mediating gene flow with immigration coming mostly from the continent (5.16%). Within England, most gene flow was to the west (from east 4.55% and from IOW 3.66%), whereas there was little flow to the east or to the IOW (Table 2). It is, however, worth noting that England has been more intensively sampled than the continent and the full extent of genetic diversity on mainland Europe has not been sampled. As a result, some finer substructuring may not have been recovered. This could affect the estimation of gene flow between the United Kingdom and mainland Europe and the values quoted are therefore to be taken as a relative indication of the magnitude of gene movement. Furthermore, as most continental samples are from central Europe rather than bordering the English Channel, gene flow is likely to be underestimated.
Mitochondrial genetic diversity and differentiation
A total of 26 haplotypes were identified among the 441 samples from 35 roosts. Thirty polymorphic sites (7.79%) were recorded with a transition/transversion ratio of 18.37. Higher diversity was observed in continental sites than in English roosts (mean pairwise differences: P<0.01; haplotype diversity: P<0.05; nucleotide diversity: P<0.01; Supplementary Table S2). Thus, all continental sites sequenced for more than one individual displayed several haplotypes. No continental haplotypes were shared among roosts, the exception being the French samples that shared all five haplotypes with several samples from eastern England and the IOW (Figure 4). In contrast, in England, one haplotype was shared by 322 out of the 409 English samples, and all but one English roost contained this haplotype. Furthermore, 13 out of the 28 English roosts were fixed for this haplotype. More diversity was found in the roosts of eastern England and the IOW than in western England, which displayed only six haplotypes, two of them being shared with the rest of England. Estimated ϕST was high at 0.405 and significant, indicating strong differentiation among roosts at the mtDNA level.

Phylogenetic analyses on HVII portion of mitochondrial D-loop. (a) Haplotype network. Each circle represents a haplotype and its size reflects the number of individuals that were identified for that haplotype. Haplotype FB/UK#4 was scaled down by 1.3 and haplotype UK#1 by 10 due to their high frequency. Care should be taken when interpreting the sizes of the circles, as they are likely more representative of sampling effort than they are of haplotype prevalence. Closed circles represent missing haplotypes. Haplotypes were colour-coded according to the population(s) they were identified in. Populations were defined according to both nuclear and mitochondrial analyses. (b) Neighbour-joining tree rooted with HVII from Chalinolobus tuberculatus. Nodes with less than 50% support from Maximum Likelihood or Bayesian analyses were collapsed. Support for the remaining nodes are indicated (ML/Bayesian). Haplotypes are colour coded as in (a).
Mitochondrial population structure
The sPCA investigated sequence variability in a spatial context. The two first axes had the highest eigenvalues and were therefore retained. PC-1 separated France and England from the rest of the continent, although the boundary was not sharp and a slight cline across the whole of Europe could be noticed. PC-2 created two clusters on the continent corresponding to eastern sites (Slovakia and Poland) and western sites (Italy, Switzerland and France). It also segregated the IOW from the rest of England, and eastern England from western England, although these last scores were very low, suggesting this structure is weak (Figure 3 and Supplementary figure S4).
AMOVA produced a simpler structure with the among groups variance component optimised for three groups (ϕCT=0.481): eastern continent, western continent and England (Table 3). Pairwise ϕST between the three groups (Table 4) indicated strong differentiation between England and both the eastern and western continental populations, and much weaker differentiation between the two continental populations. All pairwise differentiation tests were significant (P=0.008). The Mantel test revealed a significant correlation between genetic and geographical distances when controlling for the potential effects of geographical barriers (r=0.281, P<0.05; Supplementary Figure S3). However, physical barriers did not contribute in structuring genetic variation when controlling for geographical distances (r=0.109, P=0.170). When splitting the full data set into a continental one and an English one, no cline in genetic differentiation or effect of geographical barriers were detected on the continent or in England (continent: geographical distance: r=0.206, P=0.209, barriers: r=0.295, P=0.074; United Kingdom: geographical distance: r=0.079, P=0.118, barriers: r=0.019, P=0.175).
Phylogenetic analysis
The haplotype network displayed a highly connected star-shaped structure, indicative of rapid population expansion (Figure 4). Some division within England was observed, although the two haplotypes responsible differed by only 1 bp, indicating the weakness of this structure. The topology of the ML and Bayesian analyses demonstrated a similar pattern of shallow genealogy among haplotypes (Figure 4). Overall, there was a lack of obvious clustering or clear geographical patterns in the haplotype genealogies suggesting that all the haplotypes belong to the same clade and indicating rapid population expansion following a demographic bottleneck. This was confirmed by the mismatch analysis run on the three populations defined by the AMOVA and by FS values. The English population displayed significantly negative FS and nonsignificant sum of squared deviation and raggedness index, indicating no significant difference from a model of sudden expansion (Supplementary Table S4 and Supplementary Figure S4). The signal of population expansion was less clear for eastern continent and western continent as the mismatch analysis also yielded nonsignificant sum of squared deviation and raggedness indices (Supplementary Table S4), despite mismatch distributions more consistent with those from stable populations (Supplementary Figure S4). Furthermore, their FS values, while negative, were nonsignificant (Supplementary Table S4). Together, those results indicate that the continental populations are likely stable, whereas the English one recently expanded.
Discussion
Our analysis of the genetic structure of E. serotinus across Europe and within England revealed low but significant population genetic structure at the largest geographical scale, with English serotines being somewhat distinct from their European peers. There were different patterns of genetic partitioning within these populations, with stronger genetic homogeneity on the continent than in England, consistent with the hypothesis of reduced movement at edge population compared with core population. Despite this structuring, high admixture and assessment of gene flow among the differentiated populations indicated westward movement within England and a lesser role of the English Channel as physical barrier than initially hypothesised. Stronger differentiation in mtDNA was detected, suggesting females are more philopatric than males, as we expected from a bat, and therefore gene movement is mostly mediated by male connectivity. We discuss each finding in turn, as well as the limitations of our study and direction for further research where appropriate.
Genetic differentiation among roosts across Europe was low (FST=0.048) but significantly different from panmixia, with a pattern of isolation by distance indicating some limit to gene flow. Bats tend to be relatively mobile and levels of population structuring are often low (Moussy et al., 2013). For example, the migratory bat Nyctalus noctula displays very low levels of genetic structuring across Europe (FST=0.006) (Petit and Mayer, 1999). The fixation index we estimated for E. serotinus, while low, is relatively high for a bat and reflects values reported for non-migratory species (Moussy et al., 2013). Overall, the current evidence indicates E. serotinus is relatively sedentary.
Consistent with the departure from panmixia, bayesian and multivariate analyses of nuclear data recovered four genetic populations, geographically represented as continental Europe, east of England, west of England and the IOW. Some evidence for physical barriers to gene flow was also found. While there was weak evidence that mountains act as barriers, the sea appears to restrict gene flow to a degree, even between England and the IOW—6km. This distance is less than the mean commuting-distance/night recorded for E. serotinus (Catto et al., 1996). However, open water restricts movement for many bats (García-Mudarra et al., 2009; Moussy et al., 2013).
Despite the large geographical scale (>3000 km) and several mountain ranges, individual-based analyses of the nuclear markers failed to detect any strong substructuring within continental Europe. Similar findings have been reported in other taxa (Demont et al. 2008) and suggest genetic homogenisation over a large geographical scale and possibly high gene flow across the continent. Lack of information on the species’ mating system and movements restricts our understanding of the mechanisms driving this gene flow. However, it is worth noting that the sampling design to assess continental genetic diversity and distribution thereof was not ideal because only eight locations across the continent were sampled. The full extent of genetic diversity could thus not be recovered. Finer genetic substructuring could have be missed and further work with more intensive sampling across more locations is required to fully understand patterns of genetic partitioning across the continent.
Within England, the fixation index was as high as within continental Europe, in spite of the much smaller geographical scale, and smaller effective population sizes could contribute to the high fixation index there. All analyses of nuclear markers identified three UK populations: eastern England, western England and the IOW. This pattern of stronger genetic structure in England than on the continent had previously been reported in another bat (Flanders et al., 2009). The drivers for this relatively strong structuring are unknown, but the United Kingdom represents the most northern limit of many bat species, including E. serotinus, and this could affect individual behaviour (for example, dispersal) and population dynamics, and influence genetic structure. Lower dispersal at range edges has indeed been shown in other taxa (Travis and Dytham, 1999), primarily due to increased dispersal costs and reduced habitat density (Dytham, 2009). The identification of an IOW population can be explained by the sea restricting gene flow between the island and mainland England. However, no obvious physical or behavioural barriers, habitat fragmentation or lack of connectivity seem to explain what appears to be two partially isolated populations on the English mainland, although greater urbanisation in eastern England could potentially limit movement. An alternative explanation is that the east–west structure in England represents two catchments of gene circulation, possibly driven by limited and geographically distinct swarming/hibernation sites with ongoing gene flow through male, and to a lesser extent female, dispersal. Our finding of structure in England starkly contrasts with a recent study of the genetic structure of E. serotinus in Poland (Bogdanowicz et al., 2013). The geographical scale of our study and the Polish one are similar, and four out of the seven microsatellites they used were used here. However, Bogdanowicz et al., 2013 failed to detect any nuclear structure, recovering only one genetic population and very low differentiation among roosts (FST=0.01). Although these results differs from our English findings, they are consistent with our continental results, despite the sampling limits of our study.
Historical events, like the post-glacial recolonisation of Europe, influence the partitioning of genetic variation for some bats (Flanders et al., 2009), and the significantly differentiated eastern and western continental clusters we detected at the mitochondrial level could be due to post-glacial recolonisation from separate refugia, with the low structure in the nuclear markers reflecting high contemporary gene flow. In addition, lower mitochondrial diversity was found in England compared with mainland Europe, which, together with a unimodal mismatch distribution, could indicate past population bottlenecks before rapid population expansion in the United Kingdom. However, again, caution needs to be used in this interpretation because of the low continental sampling. Increasing the sample size and coverage on the continent and at the putative contact zone would be required to reconstruct the post-glacial history of the species and its contemporary influence on the distribution of genetic diversity.
Although significant genetic structure was found for E. serotinus, there was nonetheless high admixture, as indicated by Bayesian and multivariate analyses of nuclear markers and the low FST. This indicates strong ongoing gene flow among populations rather than lack of drift because of recent population divergence, which in turn indicates regular movement of individuals between populations, even across the English Channel. Specifically immigration from continental Europe to the species’ northern English range was inferred from nuclear data. Immigrants appear to be going mostly to the IOW and eastern England. Although eastern England is geographically close to continental Europe, the shortest distance to the IOW is ca 150 km, so it is less clear why it appears so connected to mainland Europe. However, interpretation here has to be tempered by the low number and the location of the continental samples and unfortunately no samples could be obtained from the continental borders of the English Channel. Gene flow analysis therefore only estimated movements from the overall continental population and may not reflect the true extent of gene movement, which are probably underestimated.
Within England, the low biparental differentiation between populations and the recovery of eastern mitochondrial haplotypes in the west indicates most bat movements are from east to west. Similarly, movements between the IOW and mainland England are mostly directed toward western England. This could indicate a westward expansion of the species range, consistent with the lower haplotypic and nucleotide diversity in western England. E. serotinus has indeed been mostly recorded in south eastern England and its presumed absence from far west has never been fully explained because habitat and roosting opportunities seem favourable. However, since 2004, the National Bat Monitoring Programme has reported a significant regional changes in bat distributions, with a steep increases in the southwest (Bat Conservation Trust, 2013). This is consistent with reports from local bat workers and volunteers registering E. serotinus calls in western counties on a more frequent basis, prompting them to suspect the establishment of new communities in the west (Marshall, personal communication).
Significant differentiation in the nuclear and the mitochondrial data occurred among sites at the European scale. However, estimates of genetic differentiation revealed that the level of population structure displayed by mtDNA haplotypes was 10 times higher than that inferred by nuclear markers. Although the comparison of differentiation between different markers is difficult because of different rates and modes of mutations, with high migration, as expected for vagile species like bats, mutation is likely to contribute little to differentiation (Balloux and Lugon-Moulin, 2002). Therefore, the observed difference between biparental and maternal fixation indices is probably the result of differences in effective population size for the markers and sex-biased gene flow. Conservatively assuming that sexual selection in E. serotinus is weak, the contrast in differentiation measured between the two types of markers exceeds their expected four-fold difference in effective population size. This suggests that gene flow in E. serotinus is mostly mediated by males, whereas higher differentiation among roosts for mtDNA indicates female philopatry (assuming the demography of this marker is a true reflection of population demography; Balloux, 2010), which correlates with the qualitative experience of workers studying E. serotinus across Europe (Hutson, personal communication; Harbusch and Racey, 2006). The suggestion of male-biased gene movement and female philopatry we infer is widely observed in temperate bats (Moussy et al., 2013), but further work analysing adult male movements is required to confirm this pattern.
In any case, some female dispersal occurs in E. serotinus as several mitochondrial haplotypes are shared among roosts. For example, all haplotypes found in French specimens are shared with roosts in eastern England and the IOW. The coexistence of these haplotypes within roosts could be the results of past colonisation events, and possibly the contemporary recruitment of dispersing females. Similarly, one haplotype common to eastern England was also identified in a roost in the west, consistent with female dispersal and recruitment. However, the pattern of isolation by distance found over Europe suggests limits to female dispersal, although this could also reflect local selection for different haplotypes (Arnqvist et al., 2010; Balloux, 2010). Furthermore, no haplotypes are shared among continental sites. The geographical scale involved there is much larger than within England, and it therefore appears that female dispersal occurs over subcontinental spatial scales (for example, <500 km). This is further supported by the fact that with the exception of the main English haplotype that is widely distributed, most shared haplotypes in the United Kingdom are found in neighbouring roosts. Hence, although female dispersal can occur over longer distances, most females apparently remain philopatric to their natal landscape if not to their natal roost.
Conclusion
Our findings indicated considerable gene flow on a large geographical scale in continental Europe despite the presence of physical barriers that could contribute to some level of substructuring, but further work with better sample coverage is required to confirm these patterns. Nonetheless, putative movement could contribute to large-scale circulation of EBLV-1 in Europe, maintaining the high viral prevalence recorded there (Schatz et al., 2013). This also seems to be concordant with the relatively low genetic diversity of EBLV-1 across Europe possibly promoted by its host’s movement (McElhinney et al. 2013). We also suggest that the English Channel, while apparently restricting some gene flow, does not appear to be a substantial barrier, and similar inferences have been drawn for M. daubentonii (Atterby et al., 2010), which is the host for EBLV-2, a virus found both on the continent and in the United Kingdom (Harris et al., 2006). The relative fragmentation of serotine populations in England may sufficiently alter host–virus epidemiology to explain the apparent absence of EBLV-1 in England. This explanation is consistent with a previous small-scale comparison of the genetic structure of the EBLV-1 (E. serotinus) and EBLV-2 (M. daubentonii ) hosts that revealed more structure within England in E. serotinus than in M. daubentonii (Smith et al., 2011). The cause(s) of the reduced movement of serotines within the United Kingdom remains to be established.
Data archiving
Mitochondrial haplotypes have been deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank), accession numbers: KP780844–KP780869. Microsatellite genotypes available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.665np.
Accession codes
References
Arnqvist G, Dowling DK, Eady P, Gay L, Tregenza T, Tuda M et al. (2010). Genetic architecture of metabolic rate: environment specific epistasis between mitochondrial and nuclear genes in an insect. Evolution 64: 3354–3363.
Atterby H, Aegerter JN, Smith GC, Conyers CM, Allnutt TR, Ruedi M et al. (2010). Population genetic structure of the Daubenton’s bat (Myotis daubentonii) in western Europe and the associated occurrence of rabies. Eur J Wildl Res 56: 67–81.
Balloux F . (2010). The worm in the fruit of the mitochondrial DNA tree. Heredity 104: 419–420.
Balloux F, Lugon-Moulin N . (2002). The estimation of population differentiation with microsatellite markers. Mol Ecol 11: 155–165.
Bat Conservation Trust. (2013). National Bat Monitoring Programme annual report. Available from www.bats.org.uk.
Beerli P, Felsenstein J . (2001). Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98: 4563–4568.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F . (1996-2004) GENETIX 4.05, Logiciel Sous Windows TM Pour la Genetique Des Populations. Université de Montpellier: France.
Bogdanowicz W, Lesiński G, Sadkowska-Todys M, Gajewska M, Rutkowski R . (2013). Population genetics and bat rabies: a case study of Eptesicus serotinus in Poland. Acta Chiropterol 15: 35–56.
Burland TM, Wilmer JW . (2001). Seeing in the dark: molecular approaches to the study of bat populations. Biol Rev 76: 389–409.
Catto CMC, Hutson AM, Racey PA, Stephenson PJ . (1996). Foraging behaviour and habitat use of the serotine bat (Eptesicus serotinus) in southern England. J Zool 238: 623–633.
Cover T, Hart PE . (1967). Nearest neighbor pattern classification. IEEE Trans Inf Theory 13: 21–27.
Demont M, Blanckenhurn WU, Hosken DJ, Garner TWJ . (2008). Molecular and quantitative genetic differentiation across Europe in yellow dung flies. J Evol Biol 21: 1492–1503.
Dietz C, Von Helversen O, Dietmar N . (2009). Serotine Bat. In: Bats of Britain, Europe and Northwest Africa. A&C Black Publishers Ltd: London, UK. p 400.
Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Duran C et al. (2011). Geneious v5.4. Available from http://www.geneious.com/.
Dytham C . (2009). Evolved dispersal strategies at range margins. Proc R Soc B Biol Sci 276: 1407–1413.
Earl DA, VonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 359–361.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Excoffier L, Smouse PE . (1994). Using allele frequencies and geographic subdivision to reconstruct gene trees within a species: molecular variance parsimony. Genetics 136: 343–359.
Excoffier L, Smouse PE, Quattro JM . (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes - Application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Flanders J, Jones G, Benda P, Dietz C, Zhang SY, Li G et al. (2009). Phylogeography of the greater horseshoe bat, Rhinolophus ferrumequinum: contrasting results from mitochondrial and microsatellite data. Mol Ecol 18: 306–318.
Fu YX, Li WH . (1993). Statistical tests of neutrality of mutations. Genetics 133: 693–709.
Furmankiewicz J, Altringham JD . (2007). Genetic structure in a swarming brown long-eared bat (Plecotus auritus) population: evidence for mating at swarming sites. Conserv Genet 8: 913–923.
García-Mudarra JL, Ibáñez C, Juste J . (2009). The Straits of Gibraltar: barrier or bridge to Ibero-Moroccan bat diversity? Biol J Linn Soc 96: 434–450.
Goudet J . (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Goudet J . (2001). FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www2.unil.ch/popgen/software.
Goudet J, Raymond M, de Meeüs T, Rousset F . (1996). Testing differentiation in diploid populations. Genetics 144: 1933–1940.
Harbusch C . (2003). Aspects of the ecology of serotine bats (Eptesicus serotinus, Schreber 1774) in contrasting landscapes in southwest Germany and Luxembourg. PhD Thesis, University of Aberdeen.
Harbusch C, Racey PA . (2006). The sessile serotine: the influence of roost temperature on philopatry and reproductive phenology of Eptesicus serotinus (Schreber, 1774) (Mammalia: Chiroptera). Acta Chiropterol 8: 213–229.
Harris SL, Brookes SM, Jones G, Hutson AM, Racey PA, Aegerter JN et al. (2006). European bat lyssaviruses: distribution, prevalence and implications for conservation. Biol Conserv 131: 193–210.
Harris SL, Aegerter JN, Brookes SM, McElhinney LM, Jones G, Smith GC et al. (2009). Targeted surveillance for European bat lyssaviruses in English bats (2003-06). J Wildl Dis 45: 1030–1041.
Hutterer R, Ivanova T, Meyer-Cords C, Rodrigues L . (2005) Bat Migrations in Europe- A Review of Banding Data and Literature. Federal Agency for Nature Conservation: Bonn, Germany.
Jombart T . (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405.
Jombart T, Devillard S, Balloux F . (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11: 94.
Jombart T, Devillard S, Dufour A-B, Pontier D . (2008). Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101: 92–103.
Jombart T, Pontier D, Dufour A-SB . (2009). Genetic markers in the playground of multivariate analysis. Heredity 102: 330–341.
Kimura M . (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120.
Kunz TH, Braun de Torrez E, Bauer D, Lobova T, Fleming TH . (2011). Ecosystem services provided by bats. Ann N Y Acad Sci 1223: 1–38.
Kuzmin IV, Bozick B, Guagliardo SA, Kunkel R, Shak JR, Tong S et al. (2011). Bats, emerging infectious diseases, and the rabies paradigm revisited. Emerg Health Threats J 4: 7159.
McElhinney LM, Marston DA, Leech S, Freuling CM, van der Poel WH, Echevarria J et al. (2013). Molecular epidemiology of bat lyssaviruses in Europe. Zoonoses Public Health 60: 35–45.
Metheny JD, Kalcounis-Rueppell MC, Willis CKR, Kolar KA, Brigham RM . (2007). Genetic relationships between roost-mates in a fission–fusion society of tree-roosting big brown bats (Eptesicus fuscus). Behav Ecol Sociobiol 62: 1043–1051.
Moran P . (1948). The interpretation of statistical maps. J R Stat Soc Ser B 10: 243–251.
Moran P . (1950). Notes on continuous stochastic phenomena. Biometrika 37: 17–23.
Moussy C, Hosken DJ, Mathews F, Smith GC, Aegerter JN, Bearhop S . (2013). Migration and dispersal patterns of bats and their influence on genetic structure. Mamm Rev 43: 183–195.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Petit E, Balloux F, Goudet J . (2001). Sex-biased dispersal in a migratory bat: a characterization using sex-specific demographic parameters. Evolution 55: 635–640.
Petit E, Mayer F . (1999). Male dispersal in the noctule bat (Nyctalus noctula): where are the limits? Proc Biol Sci 266: 1717–1722.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
R Core Team. (2014) R: a Language and Environement for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria http://www.R-project.org/.
Racey PA, Entwistle AC . (2003). Conservation ecology of bats. In: Kunz TH, Fenton B (eds) Bat Ecology. The University of Chicago Press: Chicago, USA. pp 680–743.
Rambaut A, Drummond A . (2009). Tracer v1.5. Available from http://beast.bio.ed.ac.uk/software/tracer/.
Rice W . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Robinson MF, Stebbings RE . (1997). Home range and habitat use by the serotine bat, Eptesicus serotinus, in England. J Zool 243: 117–136.
Ronquist F, Huelsenbeck JP . (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Rousset F . (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Rousset F . (2008). Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8: 103–106.
Sambrook J, Russel DW . (2001) Molecular Cloning: A Laboratory Manual third edition 3 volume set Cold Spring Harbor Laboratory Press: Cold Spring Harbor.
Schatz J, Fooks AR, McElhinney L, Horton D, Echevarria J, Vázquez-Moron S et al. (2013). Bat rabies surveillance in Europe. Zoonoses Public Health 60: 22–34.
Schneider S, Excoffier L . (1999). Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152: 1079–1089.
Smith GC, Aegerter JN, Allnutt TR, MacNicoll AD, Learmount J, Hutson AM et al. (2011). Bat population genetics and lyssavirus presence in Great Britain. Epidemiol Infect 139: 1463–1469.
Teacher AGF, Griffiths DJ . (2011). HapStar: automated haplotype network layout and visualization. Mol Ecol Resour 11: 151–153.
Travis JMJ, Dytham C . (1999). Habitat persistence, habitat availability and the evolution of dispersal. Proc Biol Sci 266: 723–728.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wilkinson G, Chapman A . (1991). Length and sequence variation in evening bat D-loop mtDNA. Genetics 128: 607–617.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Wu M, Eisen JA . (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol 9: R151.
Acknowledgements
We acknowledge DEFRA and University of Exeter for funding this study. We are grateful to the local bat groups for their help in locating and arranging access to roosts, to all the home-owners for their hospitality and tolerance during field work, and to the volunteers who assisted in the capture and sampling. We also thank Laurent Arthur, Philippe Christé, Adrino Martinoli, Martina Spada, Martin Celuch, Wieslaw Bogdanowicz and Loseb Natradze for providing the continental wing biopsies. Javier Juste shared his experience in sequencing serotine mtDNA and provided primers sequences and conditions. We also thank the editor and referee’s for constructive comments, which greatly improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Moussy, C., Atterby, H., Griffiths, A. et al. Population genetic structure of serotine bats (Eptesicus serotinus) across Europe and implications for the potential spread of bat rabies (European bat lyssavirus EBLV-1). Heredity 115, 83–92 (2015). https://doi.org/10.1038/hdy.2015.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.20
This article is cited by
-
Inferring genetic structure when there is little: population genetics versus genomics of the threatened bat Miniopterus schreibersii across Europe
Scientific Reports (2023)
-
Population structure, gene flow and relatedness of Natterer’s bats in Northern England
Mammalian Biology (2021)
-
Characterising the relationship between suitable habitat and gene flow for Myotis bechsteinii and Eptesicus serotinus in Britain
Landscape Ecology (2021)
-
Population genetic structure based on mitochondrial DNA analysis of Ikonnikov’s whiskered bat (Myotis ikonnikovi—Chiroptera: Vespertilionidae) from Korea
Journal of Ecology and Environment (2019)
-
Genetic structure and diversity of a rare woodland bat, Myotis bechsteinii: comparison of continental Europe and Britain
Conservation Genetics (2018)
