
Abstract
There is great uncertainty about how Ireland attained its current fauna and flora. Long-distance human-mediated colonization from southwestern Europe has been seen as a possible way that Ireland obtained many of its species; however, Britain has (surprisingly) been neglected as a source area for Ireland. The pygmy shrew has long been considered an illustrative model species, such that the uncertainty of the Irish colonization process has been dubbed ‘the pygmy shrew syndrome’. Here, we used new genetic data consisting of 218 cytochrome (cyt) b sequences, 153 control region sequences, 17 Y-intron sequences and 335 microsatellite multilocus genotypes to distinguish between four possible hypotheses for the colonization of the British Isles, formulated in the context of previously published data. Cyt b sequences from western Europe were basal to those found in Ireland, but also to those found in the periphery of Britain and several offshore islands. Although the central cyt b haplotype in Ireland was found in northern Spain, we argue that it most likely occurred in Britain also, from where the pygmy shrew colonized Ireland as a human introduction during the Holocene. Y-intron and microsatellite data are consistent with this hypothesis, and the biological traits and distributional data of pygmy shrews argue against long-distance colonization from Spain. The compact starburst of the Irish cyt b expansion and the low genetic diversity across all markers strongly suggests a recent colonization. This detailed molecular study of the pygmy shrew provides a new perspective on an old colonization question.
Similar content being viewed by others


Introduction
The fauna and flora of Ireland might be expected to be very similar to that of Britain. They are two large islands in an offshore archipelago separated by only a narrow gap of water. However, there are distinctly fewer species on Ireland and among those species that do occur some are absent or poorly represented in Britain, and instead are associated with southwestern Europe, known as the ‘Lusitanian element’ (Corbet, 1961; Moore, 1987). This is strikingly illustrated by small- and medium-sized mammals. Widespread and common species on Britain such as the field vole (Microtus agrestis), common shrew (Sorex araneus), mole (Talpa europea) and weasel (Mustela nivalis) are not found on Ireland and two of the species that are found on Ireland (the pygmy shrew Sorex minutus and pine marten Martes martes) have genetic affiliations to populations in southwestern Europe (Davison et al., 2001; Mascheretti et al., 2003).
There has been an intense debate about the presence–absence and relative role of in situ glacial refugia, land bridges and human introductions to explain the fauna and flora of Ireland (Davenport et al., 2008). No species has sparked more interest in this debate than the pygmy shrew (Devoy et al., 1986; Woodman et al., 1997; Yalden, 1999; Searle, 2008). There have been suggestions for a natural arrival of the pygmy shrew over land bridges from the north and from the south, and human introductions from Britain, southwestern Europe and beyond (Searle, 2008). This single species seems to encapsulate all possible hypotheses on the origin of the Irish fauna and flora. Although we know that overcoming the ‘pygmy shrew syndrome’ (Devoy et al., 1986) will not solve all issues relating to the colonization of Ireland, it is undoubtedly an important piece in the puzzle. The species is poorly represented in the fossil record of Ireland (McCormick, 1999), so contemporary genetic data appear to be the only way to untangle the colonization history of this enigmatic species in Ireland.
For the pygmy shrew there are microsatellite data (McDevitt et al., 2009) and molecular sequence data available for the mitochondrial cytochrome (cyt) b gene (Mascheretti et al., 2003; Searle et al., 2009; Vega et al., 2010a, 2010b), the mitochondrial control region (McDevitt et al., 2009, 2010) and Y chromosome introns (McDevitt et al., 2010; Vega et al., 2010a), collected from different parts of Europe to examine various phylogeographic questions. However, not since the study by Mascheretti et al. (2003) has there been a molecular analysis specifically addressing the colonization source of the Irish pygmy shrew. The purpose of the present article is to revisit this question. We utilized new data for all the molecular markers previously studied in pygmy shrews and analyzed them in the context of published results, making this study a comprehensive expansion compared with Mascheretti et al. (2003).
As already established by Mascheretti et al. (2003), there are two cyt b lineages in the British Isles: one in Ireland, which is also found in the Pyrenees (referred to as the ‘western’ lineage, to represent its European range; McDevitt et al., 2010) and another found in Britain with a northerly distribution that extends eastward as far as central Siberia (referred to as the ‘northern’ lineage; McDevitt et al., 2010). Recent data have shown that the western lineage is not just limited to Ireland and the Pyrenees; it is also found around the western and northern periphery of Britain, including the Outer Hebrides and Orkney, archipelagos off the coast of Scotland (Figure 1; Searle et al., 2009). Generalizing from results for a range of species, Searle et al. (2009) suggested that the western lineage was the first to colonize Britain, while the northern lineage colonized later, largely replacing the western lineage and leaving it in its current peripheral distribution. This pattern, as applied to all the species, was dubbed a ‘Celtic fringe’, because of the resemblance to the distribution of Celtic people. Moreover, the fact that the pygmy shrew occurs on Ireland, the Outer Hebrides and Orkney, suggests that this species, unlike the others considered by Searle et al. (2009), may have a particular tendency to be transported by humans. Ireland, the Outer Hebrides and Orkney are three island groups that may never have been attached by land to Britain or continental Europe after the end of the Last Glacial Maximum (Yalden, 1999; Searle, 2008). Hence, it has been suggested that pygmy shrews could have arrived to these places as human stowaways (Churchfield and Searle, 2008a).

Possible routes of colonization of the British Isles by the pygmy shrew in relation to the current distribution of the western (grey) and northern (black) cyt b lineages shown schematically in a2a, a2b, b2a and b2b. Panel a1 represents the first stage in the colonization scenarios by a western lineage in a2a and a2b, and panel b1 by a northern lineage for scenarios b2a and b2b. The maps show continental Europe with Britain and the more westerly Ireland as the large offshore islands, and the Outer Hebrides and Orkney (labelled OH and ORK, respectively in a1). Arrows originating from the bottom of panels represent Iberia. The current peripheral distribution of the western lineage in Britain is shown in a highly stylized way in a2a, a2b, b2a and b2b. Actually, the northern lineage tends to be present right up to the western coast and occurs as a polymorphism with the western lineage there. So, in the Celtic fringe scenarios (a2a, a2b), the colonization of islands by the western lineage from the British mainland would have had to occur before the invading northern lineage became so predominant along the west coast (Searle et al., 2009).
Therefore, given what we already know about pygmy shrew cyt b variation in the British Isles from Mascheretti et al. (2003) and Searle et al. (2009), and what has been written in the literature on how the pygmy shrew colonized Ireland (Yalden, 1981, 1999; Mascheretti et al., 2003; Searle, 2008; McDevitt et al., 2009; Searle et al., 2009), we test four alternative hypotheses for the colonization of the British Isles by pygmy shrews (Figure 1). The ‘a’ hypotheses are Celtic fringe scenarios, where a1 represents the first stage of the colonization of Britain by the western lineage from continental Europe. In ‘a2a’, Ireland is colonized in the same way as the other Celtic fringe islands (Orkney, Outer Hebrides), directly from the British mainland (most likely by being stowaways on boats but alternatively over a land bridge). In ‘a2b’, the pygmy shrew is accidentally introduced onto Ireland as stowaways on boats from southwestern Europe, taking into account the similarity in the fauna and flora between Ireland and southwestern Europe. The importance of this possible long-distance source of introduction of the pygmy shrew is taken further in the ‘b’ hypotheses, which consider the western lineage as coming to Britain entirely by this route, either via Ireland to other parts of the British Isles (‘b2a’) or independently to different parts of the distribution (‘b2b’). In these ‘b’ hypotheses, the only natural colonization of Britain is by the northern lineage (b1), while the western lineage arrived with humans and replaced the northern lineage from parts of the periphery of mainland Britain.
In this paper, we will use molecular data to distinguish between these four hypotheses.
Materials and methods
Specimens
Specimens collected by us or colleagues from Britain (and its islands), Ireland, France, Spain, Norway and Sweden were subjected to molecular analysis. The specimens were preserved in ethanol and are stored at the National Museums of Scotland, Edinburgh, UK (NMSZ.2009.101). In total, we report here 218 new cyt b sequences, 153 new control region sequences, 17 new Y-intron sequences and 335 new microsatellite multilocus genotypes (Supplementary Table S1).
Molecular methods
The methods for DNA extraction and sequencing part of the cyt b gene follow Searle et al. (2009) and those for sequencing part of the control region and four Y chromosome introns (DBY1, DBY3, DBY7 and UTY11) follow McDevitt et al. (2010). The total sequence lengths used for analysis were: cyt b, 1110 base pairs (bp); control region, 342 bp; Y-introns, 2939 bp (concatenated). We used microsatellites developed in related Sorex species (Wyttenbach et al., 1997; Balloux et al., 1998; Naitoh et al., 2002). Nine loci were studied, five (A6, B4, D2, D8 and L33) according to the protocol from McDevitt et al. (2009), and four (L9, L14, L62 and L69) were amplified in 25 μl PCR reactions using the following conditions: 94 °C for 4 min, 30 cycles of 94 °C for 45 s, 55–57 °C for 45 s, 72 °C for 45 s and a final extension at 72 °C for 7 min.
Analysis
For the cyt b gene, the phylogenetic analysis was entirely focused on one particular phylogenetic group, already identified by Mascheretti et al. (2003) and Searle et al. (2009) and here named as the ‘western lineage’ (see Introduction section). A phylogenetic network was constructed using the software Network version 4.5.1.0 (http://www.fluxus-engineering.com) with a median-joining algorithm based on maximum parsimony and a ‘greedy’ method (Bandelt et al., 1999). Under the circumstances of closely related sequences, there are advantages in using a median-joining network to depict relationships (Posada and Crandall, 2001) and simulation studies have demonstrated that this method provides reliable estimates of the true genealogy (Cassens et al., 2005; Woolley et al., 2008).
We used the cyt b data set of the western lineage from different geographical areas and the northern lineage from Britain (for comparison) to determine the nucleotide and haplotype diversities and the neutrality test statistics Tajima's (1989) D, Fu's (1997) FS and Ramos-Onsins and Rozas's (2002) R2 using the program DnaSP 4.90.1 (Rozas et al., 2003). Although designed to detect selection, D, FS and R2 are indices that respond to population expansions. The significance of D was tested under the assumption that D follows the beta distribution and for FS and R2 significance was assessed by comparison with a null distribution obtained from 10 000 coalescent simulations. The maximum pairwise divergence between haplotypes within a geographical area (in terms of nucleotide substitutions) was determined using the program Arlequin 3.12 (Excoffier et al., 2005). Given the nature of the sampling (some sites with very few specimens (1 or 2) and others with many (15 or more)), the analyses were conducted on either a single individual per locality or up to five individuals per locality, with individuals randomly selected for those localities with more than five individuals.
Neighbor-joining, maximum parsimony and maximum likelihood phylogenetic trees from a subset of control region haplotypes (128, including 60 new British haplotypes; Supplementary Table S1) were constructed in PAUP* 4.0b10 (Swofford, 1998). The neighbor-joining and maximum likelihood trees were constructed using the HKY85+I+G (Hasegawa et al., 1985) model (I=0.3185; α=0.675) selected by the Akaike Information Criterion in Model Test 3.06 (Posada and Crandall, 1998). Branch support was calculated from 1000 neighbor-joining, maximum parsimony and maximum likelihood bootstrap pseuodoreplicates. Bayesian posterior probabilities were estimated in MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003) from 1 000 000 generations sampled every thousandth generation excluding a burn-in of 100 000 steps. The Caucasian pygmy shrew (Sorex volnuchini) was used as the outgroup species as in previous phylogeographic studies on S. minutus (Mascheretti et al., 2003; Searle et al., 2009; McDevitt et al., 2010; Vega et al., 2010a, 2010b).
A median-joining network of concatenated Y chromosomal introns was constructed according to the method described above for cyt b sequences. Indels were included as informative characters (McDevitt et al., 2010).
Using the genotypes obtained from the nine microsatellite loci, we examined how pygmy shrews from Ireland, Britain (and several of its islands) and Scandinavia clustered genetically. First, we used a Principal Component Analysis with the software JMP version 8.0 (SAS Institute, Cary, NC, USA). Second, we applied the Bayesian algorithm implemented in STRUCTURE v. 2.3.3 (Pritchard et al., 2000). In total, 335 individuals were included in these analyses, with the minimum criteria of being genotyped at 7 of the 9 loci (see Supplementary Table S1). Broken down, there were 157 Irish, 157 British and 21 Scandinavian individuals. For STRUCTURE, five independent runs were performed for each K value (1–20) using 500 000 iterations, with a burn-in period of 100 000 iterations. STRUCTURE was run using the default settings (Pritchard et al., 2000) with the admixture model. The number of clusters (K) was calculated by obtaining the mean posterior probability of the data (log probability of data; Ln P (K)) over the five independent runs. This methodology provided inconclusive results (see Supplementary Figure S3), so we applied the ΔK method (Evanno et al., 2005) to estimate the number of clusters based on the rate of change in the log probability of data between successive K values. Individuals were assigned to clusters based on their highest membership coefficient to a particular cluster averaged over the five independent runs.
Results
Cyt b and control region sequences
All new haplotypes were deposited in Genbank (see Supplementary Table S1). We constructed a phylogenetic network for the western cyt b lineage (Figure 2), including haplotypes distributed over a geographic range from Iberia to the British Isles (Figure 3a). There were two central haplotypes in the network, ‘A’ and ‘B’. The most basal was haplotype A, found in one individual in northern Spain. Haplotype B was found in two localities along the western coast of Britain. Within the network, there were haplotypes from Iberia and the British Isles that derived from each of the central haplotypes while haplotypes from France only derived from haplotype A.

Median-joining (MJ) network of cyt b haplotypes of the western lineage with circles representing haplotypes and lines representing mutational steps. The two central haplotypes of the network are labelled (A, B). The central haplotypes of other expansions within the network are also highlighted (see text): H, Outer Hebrides; I, Ireland; M, Orkney (Mainland Orkney+Westray+Hoy); S, Orkney (South Ronaldsay). Within the central haplotype of the Irish expansion the black segment represents Iberian specimens with the same haplotype. Other haplotypes highlighted (see text): the divergent haplotypes found in the most southeasterly samples of the western lineage on mainland Britain (labelled *), the Hoy haplotype not in the Mainland Orkney expansion (labelled +).

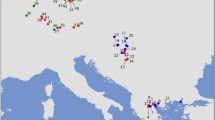
Maps showing the distribution of cyt b (a) and Y-intron (b) lineages in the British Isles in the context of continental Europe, including published data (Mascheretti et al., 2003; Searle et al., 2009; McDevitt et al., 2010; Vega et al., 2010a, 2010b). All the Irish data and the British data for Y-introns are new; likewise for some British and continental European cyt b data (see Supplementary Table S1). For the Y-intron data, the haplotypes in the British Isles are labelled according to Supplementary Table S1 and Figure 4. Only a single individual per locality was typed for the Y-introns, except one locality in Ireland where the two individuals yielded different haplotypes.
We found no sharing of haplotypes among the three geographical areas of Iberia, France and the British Isles, except for one very important exception: The central and most common haplotype in Ireland (haplotype ‘I’: Figure 2) was also found in northern Spain. This haplotype generated a clear starburst expansion in the network, which consisted solely of sequences found on Ireland (excepting the Spanish sequences mentioned above), and all sequences found on Ireland were within this starburst (Figure 2). The 94 individuals sequenced on Ireland had 35 haplotypes. Only eight haplotypes (from eight individuals) had sequences that differed by more than two substitutions from the Irish central haplotype. This pattern indicates a recent population expansion in Ireland relative to the rest of the western lineage.
There was also a well-documented (and apparently recent) expansion in the Orkney island group. Samples from the islands of Mainland Orkney, Hoy and Westray formed a group of eight related haplotypes (1–2 substitutions from the central haplotype ‘M’ in Figure 2). The full genetic diversity of this expansion was present on Mainland Orkney, where all eight haplotypes were found among 43 individuals (Supplementary Table S1). Within the group, all the individuals from Westray (33) and one from Hoy shared the same haplotype, although another individual from Hoy had a distantly related haplotype (Figure 2).
For the other Orkney island sampled, South Ronaldsay, 37 out of 40 individuals had the same haplotype (haplotype ‘S’ in Figure 2), the remaining three individuals had a closely related one. These haplotypes are phylogenetically distinct from those found on Mainland Orkney.
The pygmy shrews from the Outer Hebrides (three islands: Benbecula (one individual), North and South Uist (two individuals each)) also had three closely related haplotypes (two haplotypes with one substitution difference from a basal haplotype ‘H’) (Figure 2).
Most of the haplotypes from the British Isles derived from central haplotype B. Those that derived from central haplotype A included the three most southeastern samples within Britain (Figure 2) and one sample from the west coast. The other two samples from southeastern Britain appeared on the other side of the network, 8–9 substitutions separated from central haplotype B (Figure 2).
The measures of genetic diversity and neutrality tests for the cyt b data were little affected whether based on one individual per locality or five (Table 1). The northern lineage in Britain and the western lineage (both as a whole or broken into its geographic components) showed evidence of population expansions. The western lineage in continental Europe showed the weakest signal of population expansion based on the neutrality tests, as expected if the source of the original, earliest expansion was in the continental area. The western lineage in Ireland showed low nucleotide diversity, while the western lineage in Britain had similar nucleotide diversity to the western lineage in continental Europe and the western lineage overall, and to the northern lineage in Britain. Measures of pairwise divergence also showed that there were highly divergent haplotypes within the western lineage in Britain (Table 1), supporting observations from the cyt b network (see above, Figure 2). Overall this phylogeographic pattern was consistent with an early colonization of Britain by two waves of expansions that originated in continental Europe, so that Britain carries the full diversity of each lineage. However, the phylogeographic pattern of Ireland indicated a more recent colonization and population expansion with little genetic diversification.
The cyt b phylogeny and distribution of lineages was replicated precisely by the control region sequence data (Supplementary Figures S1 and S2), as expected given the physical linkage within the mitochondrial genome, but regardless of the higher mutation rate (McDevitt et al., 2009).
Y chromosome intron sequences
The concatenated Y chromosome intron sequences that we obtained are available on request from the investigators. As expected from previous studies using such sequences, we identified a smaller number of Y-intron lineages in continental Europe compared with those for mitochondrial DNA sequences (McDevitt et al., 2010). Nevertheless, there were clear western and northern Y-intron lineages (Figures 3b and 4). The western Y-intron lineage was found in the British Isles, France, Andorra and Spain, all places occupied by the western cyt b lineage. However, while both the western and northern cyt b lineages were found in the British Isles, only the western Y-intron lineage was found there. The three Irish Y-intron haplotypes formed a monophyletic clade but the most basal of these (IE1) was only separated by a single substitution from the most basal British haplotype (GB1) (Figure 4). GB1 was also the most basal sequence in the western Y-intron lineage (Figure 4) and was the most common and widespread haplotype in Britain (Figure 3b). Similarly, IE1 was the most common and widespread Y-intron haplotype in Ireland. None of the Y-intron haplotypes found in the British Isles have been found elsewhere in Europe.

Median-joining (MJ) network of concatenated Y-intron haplotypes colored by lineage. Numbers on branches indicate more than one mutation event. Haplotypes found in Britain and Ireland are labelled GB and IE, respectively (see Figure 3b). See McDevitt et al. (2010) for sampling locations of continental European haplotypes.
Microsatellites
A total of 166 alleles were found from 9 microsatellite loci in 335 individuals. Of these alleles, 102 were found in Ireland (157 individuals) with only 4 alleles unique there (each occurring only once). On the other hand, a total of 149 alleles were found in Britain (157 individuals), with 44 unique alleles. A further 11 alleles were unique to Scandinavia (21 individuals).
Figure 5 shows the clustering of individuals after a Principal Component Analysis, according to their geographical origin. The first two components explained 30.4% of the total variation. Individuals from Britain, Ireland and Scandinavia form three largely separate clusters, but with the British cluster overlapping the others, more so with the Irish cluster. The clustering analysis in STRUCTURE was largely inconclusive until the ΔK method (Evanno et al., 2005) was applied (Supplementary Figure S3). This revealed the presence of two distinct clusters (Figure 6). Irish shrews had a clear tendency to be assigned to Cluster 1 (light grey) while the British and Scandinavian shrews largely belonged to Cluster 2 (dark grey) but with a greater Cluster 1 presence in Britain.

Principal Component Analysis of the microsatellite data, showing each individual classified according to the geographical area in which it occurred.

Graphical output from Bayesian analysis in STRUCTURE showing assignment probabilities to Clusters 1 (light grey) and 2 (dark grey). ‘IE’ represents individuals from Ireland; ‘GB’ Britain and ‘SCA’ Scandinavia.
Discussion
Mascheretti et al. (2003) proposed a colonization history of the Irish pygmy shrew based on 18 cyt b sequences from the British Isles and 2 cyt b (western lineage) sequences from southwestern Europe. Although they established an important similarity between the sequences in Ireland and southwestern Europe, which resembled a Lusitanian distribution (Corbet, 1961), their inferences were hampered by limited sampling. In this study, we used 354 cyt b sequences from the British Isles and 20 cyt b sequences (western lineage) from southwestern Europe (France, Andorra and Spain), in addition to control region and Y-intron sequences and microsatellites. So, we believe that we are in a much stronger position to infer the uncertain source area for the colonization of Ireland.
Colonization routes of the British Isles
In Figure 1, we presented four different hypotheses for the colonization of Ireland in the context of wider colonization scenarios of the British Isles. Of these scenarios our data would appear to rule out b2a, rejecting a long-distance colonization of Ireland and subsequent colonization of the west coast of Britain from Ireland to explain the peripheral distribution of the western lineage in Britain. Our cyt b and control region phylogenies (Figure 2, Supplementary Figure S1) showed that the mitochondrial lineage found on Ireland was a distinct lineage from the rest of the British Isles. In addition, the genetic diversity of Ireland was smaller than that of Britain, and the starburst pattern of the Irish haplotypes was much more compact than that of the haplotypes from Britain (which had larger number of mutations among haplotypes), indicating a more recent origin of the Irish population (McDevitt et al., 2009). Therefore, Ireland could not be the source of other populations in the British Isles.
The other ‘b’ scenario (b2b) also looks unlikely, particularly in the light of the data from the Y-introns (Figures 3b and 4). All individuals examined from Britain belonged to the western lineage for this male-transmitted marker. Surely it is most reasonable to suggest that the presence of a male western Y-intron lineage in Britain represents the same colonization event that brought the female western cyt b lineage from continental Europe across a land bridge after the Last Glacial Maximum. It is believed that the male and female western lineages both derived from the same glacial refugium (McDevitt et al., 2010) and the colonization of Britain would have been part of the postglacial expansion from that refugium. Under these circumstances, the western lineages would have been the first colonizers of Britain, and the northern cyt b lineage (that derived from a different glacial refugium: McDevitt et al., 2010; Vega et al., 2010b) would have come in later, partially replacing the western cyt b lineage. This was perhaps as a selective sweep specific to the mitochondrial genome (Searle et al., 2009), because the Y-intron lineage that apparently derived from the same northern glacial refugium (McDevitt et al., 2010) did not penetrate Britain. In other words, in light of the Y-intron data, the ‘a’ scenarios for the colonization of Britain (Figure 1) appear more likely than b2b; there is no need to invoke multiple long-distance human introductions by boat from Iberia to explain the presence of the western cyt b lineage around the periphery of Britain. The ‘a’ scenarios are also favored for other species (bank vole Myodes glareolus, field vole and water vole Arvicola terrestris), which all have a peripheral mitochondrial DNA lineage in Britain (a ‘Celtic fringe’) and no suggestion of an influence of human introduction (Searle et al., 2009).
There are other reasons for doubting the b2b scenario. If there were multiple human introductions from Iberia to populate the periphery of Britain and surrounding islands with the western cyt b lineage, then there is no clear expectation that the island populations off Britain would be derived from the British mainland. But the molecular data indicate precisely that, in line with the ‘a’ scenarios (Figure 1). Pygmy shrews on both the Outer Hebrides and Orkney are characterized by the Y-intron haplotype GB1 that is the most common and widespread throughout mainland Britain (Figure 3b) and the central British haplotype within the Y-intron network (Figure 4). Similarly, both the Outer Hebrides and Orkney were colonized by cyt b haplotypes just one substitution removed from the central western lineage haplotype B present in Britain, with subsequent local diversification (Figure 2).
Therefore, the genetic data presented here support the ‘a’ scenarios, and there are different facets of the data that appear to favor a2a or a2b. The Y-intron data fit better with scenario a2a. The central Irish haplotype differed by just a single nucleotide substitution from the central haplotype found in Britain (Figure 4). Other haplotypes with only one substitution from the central haplotype were found in Britain, so it appears reasonable that the Irish central haplotype arose in Britain and colonized Ireland from there. A similar scenario has been suggested to explain the single nucleotide difference between mitochondrial control region lineages in house mice on Madeira (large island) and Porto Santo (nearby smaller island), again with the ancestral lineage found on the presumptively first-colonized larger island (Gündüz et al., 2001).
The microsatellite data are consistent with the scenario a2a; Ireland has essentially a subset of the alleles that occur in Britain, with a mere four unique alleles found. In the STRUCTURE analysis, this depleted diversity on Ireland was marked by a separate cluster, yet still showed admixture with British individuals (Figure 6). In the Principal Component Analysis, there were three clusters corresponding to Ireland, Britain and Scandinavia, and again the shrews from Britain showed greater similarity to those from Ireland (Figure 5). Previously published allozyme data also indicated genetic similarity between pygmy shrews in Britain and Ireland (Searle, 1989).
Long-distance colonization of Ireland from southwestern Europe (scenario a2b) is still a possibility, and the occurrence of exactly the same cyt b haplotype in northern Spain as the most prevalent one in Ireland, reinforces this possibility. Nevertheless, although the shared Spanish-Irish cyt b haplotype has not yet been found in Britain, it is reasonable to consider that it was (and still could be) present there, given the substantial cyt b variation in Britain (Table 1). With the exception of this shared haplotype between Ireland and northern Spain, the haplotypes found in Ireland are more closely associated with the majority of haplotypes found in Britain (Figure 2).
Overall then we favor scenario a2a, but our molecular results do not entirely rule out scenario a2b. Given the very low density of pygmy shrews in southern Europe (Vega et al., 2010a), it would not be easy to collect large samples of the species from northern Iberia, which may help us to firmly decide between a2a and a2b. Nevertheless, we believe that we can choose between those two scenarios now, based on available evidence from the field biology of the pygmy shrew and its distribution in the North Atlantic region.
The distributional differences within the British Isles between the pygmy and common shrew are particularly informative. The pygmy shrew is found on Ireland, Isle of Man, Orkney and the Outer Hebrides, while the common shrew is absent from them; as is also true, in general, for other non-commensal species of small mammal (Harris and Yalden, 2008). Humans apparently introduced small mammals onto these islands, and pygmy shrews were able to colonize and common shrews were not (Churchfield and Searle, 2008a). This matches well with the fact that the pygmy shrew, being smaller, has lower energy needs than the common shrew, and therefore could survive longer in a boat with small quantities of insect food, and that pygmy shrews are better climbers than common shrews and through its reduced size can get into places that a common shrew is unable to, making them more likely to get on to the boat in the first place and to be better at searching for food once on it (Churchfield and Searle, 2008a, 2008b). Also, pygmy shrews would appear more likely than common shrews to be transported in hay shipments for livestock; they are more likely to be in any grass getting cut because they are more surface-living than the common shrew (Croin Michielsen, 1966) and are more likely to climb into hay stooks. There has been considerable movement of livestock (and therefore their fodder) between mainland Britain and surrounding islands since Mesolithic times (Corbet, 1961; Yalden, 1999; Rowley-Conwy, 2004).
Both scenarios a2a and a2b involve the human introduction of pygmy shrews on to Ireland. The difference is the distance travelled in making that introduction. For a2a, this distance would have been of the same magnitude as the other islands around the British Isles onto which pygmy shrews were introduced. However, there may be limits on how far the pygmy shrew can be transported, which would make the journey from Iberia to Ireland, as required for scenario a2b, to be unrealistic. The pygmy shrew is not found on Shetland or Faroe, which are much closer to mainland Britain than the distance between northern Spain and Ireland. Nor is the species found on Iceland, despite considerable traffic of vessels from areas where pygmy shrews are found (British Isles and Scandinavia) over the last thousand years. Pygmy shrews have an extremely high metabolic rate (Taylor, 1998) and may be unable to sustain themselves on the limited insect food likely to be available in a vessel over a very long voyage. Shetland is 175 km from the nearest point on mainland, and 100 km from the nearest Orkney island with pygmy shrews; Faroe and Iceland are further from landmasses with pygmy shrews; and the journey distance between northern Spain and Ireland is 1000 km. Ireland, Orkney and the Outer Hebrides, all islands with pygmy shrews, are within 25 km of mainland Britain or other large offshore islands with pygmy shrews.
Implications for ‘The Irish Question’
What implications do our results with pygmy shrews have on our understanding of the colonization of Ireland by animals and plants (Moore, 1987)? We believe that the pygmy shrew provides an exemplary case of a human introduction onto Ireland. Our results make no demands on the presence of a land bridge between Ireland and either Britain or continental Europe after the Last Glacial Maximum. On grounds of faunistics and geophysical studies, there is a severe doubt as to whether there was a land bridge between Ireland and Britain (Lambeck and Purcell, 2001; Searle, 2008). The genetic evidence provided here plus the additional biological and distributional data suggest that the species may readily be transported short distances with humans and fits well with McDevitt et al.'s (2009) molecular analysis dating the Irish arrival of the pygmy shrew to the Holocene. This is a period when Mesolithic or (more likely) Neolithic people (McDevitt et al., 2009) could have made the introduction (before the Holocene, humans were not present on Ireland; Woodman et al., 1997). However, the pygmy shrew is a species that has long been viewed as a ‘native’ of Ireland (for example, Yalden, 1981) and this is not an easy notion to dispel. Extending our findings with pygmy shrews to other species, we recommend that care should be taken before suggesting natural colonization of Ireland for organisms that do not disperse well through the air or over a marine barrier or which are not cold-tolerant.
In considering human introduction onto Ireland, our results show that it is important to propose alternative hypotheses for possible source areas. In particular, we showed that it is critical to sample Britain carefully before ruling it out as a source area for Irish colonization of a particular species, even when there appears to be a strong genetic affiliation between Ireland and southwestern Europe (Davison et al., 2001; Mascheretti et al., 2003). The model of Corbet (1961) of introductions to Ireland directly from southwestern Europe to explain the Lusitanian distribution of certain species (or genetic types) is appealing; but we provide another explanation for presence of southwestern European forms on Ireland, that is, human-mediated colonization via Britain. Previous explanations for the occurrence of southwestern European forms on Ireland are potentially based on replacement events in Britain, certainly in the case of the pygmy shrew here. Genetic and species distributions may be dynamic over time and current genetic patterns could be misleading as a signal of the past, particularly if areas or regions are sampled insufficiently. Overall, we believe that resolving the ‘pygmy shrew syndrome’ is an important part in understanding the manner in which species colonized Ireland.
References
Balloux F, Ecoffey E, Fumagalli L, Goudet J, Wyttenbach A, Hausser J (1998). Microsatellite conservation, polymorphism, and GC content in shrews of the genus Sorex (Insectivora, Mammalia). Mol Biol Evol 15: 473–475.
Bandelt HJ, Forster P, Rohl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Cassens I, Mardulyn P, Milinkovitch MC (2005). Evaluating intraspecific ‘network’ construction methods using simulated sequence data: do existing algorithms outperform the Global Maximum Parsimony approach? Syst Biol 54: 363–372.
Churchfield S, Searle JB (2008a). Pygmy shrew. In: Harris S, Yalden DW (eds) Mammals of the British Isles Handbook 4th edn. The Mammal Society: London. pp 267–271.
Churchfield S, Searle JB (2008b). Common shrew. In: Harris S, Yalden DW (eds) Mammals of the British Isles Handbook 4th edn, The Mammal Society: London. pp 257–265.
Corbet GB (1961). Origin of the British insular races of small mammals and of the ‘Lusitanian’ fauna. Nature 191: 1037–1040.
Croin Michielsen N (1966). Intraspecific and interspecific competition in the shrews Sorex araneus L. and S. minutus L. Arch Néerland Zool 17: 73–174.
Davenport JL, Sleeman DP, Woodman PC (2008). Mind the Gap: Postglacial Colonization of Ireland. Irish Naturalists’ Journal Special Supplement: Dublin.
Davison A, Birks JDS, Brookes RC, Messenger JE, Griffiths HI (2001). Mitochondrial phylogeography and population history of pine martens Martes martes compared with polecats Mustela putorius. Mol Ecol 10: 2479–2488.
Devoy RJN, Sleeman DP, Woodman PC (1986). Conclusions. In: Sleeman DP, Devoy RJN, Woodman PC (eds). Proceedings of the Postglacial Colonization Conference. Occasional Publications of the Irish Biogeographical Society: Dublin, Vol. 1, pp 86–88.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics Online 1: 47–50.
Fu Y-X (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Gündüz I, Auffray J-C, Britton-Davidian J, Catalan J, Ganem G, Ramalhinho MG et al. (2001). Molecular studies on the colonization of the Madeiran archipelago by house mice. Mol Ecol 10: 2023–2029.
Harris S, Yalden DW (2008). Mammals of the British Isles Handbook 4th edn. The Mammal Society: London.
Hasegawa M, Kishino K, Yano T (1985). Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174.
Lambeck K, Purcell AP (2001). Sea-level change in the Irish Sea since the Last Glacial Maximum: constraints from isostatic modelling. J Quaternary Sci 16: 497–506.
McCormick F (1999). Early evidence for wild animals in Ireland. In: Benecke N (ed). The Holocene History of the European Vertebrate Fauna: Modern Aspects of Research. Verlag Marie Leidorf GmbH: Rahden, Germany. pp 355–371.
McDevitt AD, Rambau RV, O’Brien J, McDevitt CD, Hayden TJ, Searle JB (2009). Genetic variation in Irish pygmy shrews Sorex minutus (Soricomorpha: Soricidae): implications for colonization history. Biol J Linn Soc 97: 918–927.
McDevitt AD, Yannic G, Rambau RV, Hayden TJ, Searle JB (2010). Postglacial recolonization of continental Europe by the pygmy shrew (Sorex minutus) inferred from mitochondrial and Y chromosomal DNA sequences. In: Habel JC, Assman T (eds). Relict Species: Phylogeography and Conservation Biology. Springer Verlag: Heidelberg, Berlin. pp 217–236.
Mascheretti S, Rogatcheva MB, Gündüz I, Fredga K, Searle JB (2003). How did pygmy shrews colonize Ireland? Clues from a phylogenetic analysis of mitochondrial cytochrome b sequences. Proc R Soc Biol Sci B 270: 1593–1599.
Moore PD (1987). Snails and the Irish question. Nature 328: 381–382.
Naitoh Y, Ishibashi Y, Abe S, Ohdachi SD (2002). Isolation and characterization of polymorphic microsatellite DNA markers in two shrew species, Sorex unguiculatus and S. caecutiens. Mol Ecol Notes 2: 434–436.
Posada D, Crandall KA (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817–818.
Posada D, Crandall KA (2001). Intraspecific phylogenetics: trees grafting into networks. Trends Ecol Evol 16: 37–45.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Ramos-Onsins SE, Rozas J (2002). Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19: 2092–2100.
Ronquist F, Huelsenbeck JP (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Rowley-Conwy P (2004). How the west was lost: a reconsideration of agricultural origins in Britain, Ireland, and Southern Scandinavia. Curr Anthropol 45: S83–S113.
Rozas J, Sánchez-Del Barrio JC, Messeguer X, Rozas R (2003). DNAsp, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497.
Searle JB (1989). Genetic studies of small mammals from Ireland and the Isle of Man. Irish Nat J 23: 112–113.
Searle JB (2008). The colonization of Ireland by mammals. In: Davenport JL, Sleeman DP, Woodman PC (eds). Mind the Gap: Postglacial Colonization of Ireland. Irish Naturalists' Journal Special Supplement: Dublin, pp 109–115.
Searle JB, Kotlík P, Rambau RV, Marková S, Herman JS, McDevitt AD (2009). The Celtic fringe of Britain: insights from small mammal phylogeography. Proc R Soc B Biol Sci B 276: 4287–4294.
Swofford DL (1998). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10. Sinauer Associates: Sunderland, MA.
Tajima F (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Taylor JRE (1998). Evolution of energetic strategies in shrews. In: Wójcik JM, Wolsan M (eds). Evolution of Shrews. Mammal Research Institute, Polish Academy of Sciences: Białowieża, Poland. pp 309–346.
Vega R, Amori G, Aloise G, Celleni S, Loy A, Searle JB (2010a). Genetic and morphological variation in a Mediterranean glacial refugium: evidence from Italian pygmy shrews, Sorex minutus (Mammalia, Soricomorpha). Biol J Linn Soc 100: 774–787.
Vega R, Fløjgaard C, Lira-Noriega A, Nakazawa Y, Svenning J-C, Searle JB (2010b). Northern glacial refugia for the pygmy shrew Sorex minutus in Europe revealed by phylogeographic analyses and species distribution modelling. Ecography 33: 260–271.
Woodman P, McCarthy M, Monaghan N (1997). The Irish Quaternary Fauna Project. Quaternary Sci Rev 16: 129–159.
Woolley SM, Posada D, Crandall KA (2008). A comparison of phylogenetic network methods using computer simulation. PLoS One 3: e1913.
Wyttenbach A, Favre L, Hausser J (1997). Isolation and characterization of simple sequence repeats in the genome of the common shrew. Mol Ecol 6: 797–800.
Yalden DW (1981). The occurrence of the pygmy shrew Sorex minutus on moorland, and the implications for its presence in Ireland. J Zool 195: 147–156.
Yalden DW (1999). The History of British Mammals. Poyser: London.
Acknowledgements
We are grateful to all individuals who donated specimens, in particular Karl Fredga, Natália Martínková, Tom White and Damien McDevitt, who provided critical material. We thank the three anonymous referees for important and constructive comments on the paper. ADM was funded by an Embark Postgraduate Scholarship from the Irish Research Council for Science, Engineering and Technology (IRCSET) and a ULYSSES travel grant to France. RV was supported by CONACyT (México; Reg. No. 181844). RVR received funds from the Skye Foundation and the South African National Research Foundation. GY was awarded a grant from the Fondation Agassiz (Switzerland).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
McDevitt, A., Vega, R., Rambau, R. et al. Colonization of Ireland: revisiting ‘the pygmy shrew syndrome’ using mitochondrial, Y chromosomal and microsatellite markers. Heredity 107, 548–557 (2011). https://doi.org/10.1038/hdy.2011.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2011.41
Keywords
This article is cited by
-
Retracing the history and planning the future of the red squirrel (Sciurus vulgaris) in Ireland using non-invasive genetics
Mammal Research (2018)
-
Ancient Biological Invasions and Island Ecosystems: Tracking Translocations of Wild Plants and Animals
Journal of Archaeological Research (2018)
-
Molecular and morphological insights into the origin of the invasive greater white-toothed shrew (Crocidura russula) in Ireland
Biological Invasions (2016)
-
Revisiting the phylogeography and demography of European badgers (Meles meles) based on broad sampling, multiple markers and simulations
Heredity (2014)
-
The current distribution and potential for future range expansion of feral ferret Mustela putorius furo in Ireland
European Journal of Wildlife Research (2013)

{kind=link}
{kind=link}
{kind=link}