
Abstract
p53's apoptotic program consists of transcription-dependent and transcription-independent pathways. In the latter, physical interactions between mitochondrial p53 and anti- and pro-apoptotic members of the Bcl2 family of mitochondrial permeability regulators are central. Using isogenic cell systems with defined deficiencies, we characterize in detail how mitochondrial p53 contributes to mitochondrial permeabilization, to what extent its action depends on other key Bcl2 family members and define its release activity. We show that mitochondrial p53 is highly efficient in inducing the release of soluble and insoluble apoptogenic factors by severely disrupting outer and inner mitochondrial membrane integrity. This action is associated with wild-type p53-induced oligomerization of Bax, Bak and VDAC and the formation of a stress-induced endogenous complex between p53 and cyclophilin D, normally located at the inner membrane. Tumor-derived p53 mutants are deficient in activating the Bax/Bak lipid pore. These actions are independent of Puma and Bax. Importantly, the latter distinguishes the mitochondrial from the cytosolic p53 death pathway.
Similar content being viewed by others


Introduction
p53's apoptotic program, which is critically important for its tumor suppressor action in vivo, is pleiotropic and involves transcription-dependent nuclear and transcription-independent cytoplasmic pathways, designated as the transcriptional and mitochondrial arm of p53-mediated death (reviewed in 1, 2). The BH3-only protein Puma and to a lesser extent the BH123 protein Bax are important transcription-dependent apoptotic effectors of p53. On the other hand, the mitochondrial arm directly links the p53 protein to the intrinsic death pathway and triggers the shortest possible death circuitry of the cell. It acts via p53 complex formation with anti- as well as pro-apoptotic members of the Bcl2 family that triggers the process of mitochondrial outer membrane permeabilization (MOMP). In response to death stimuli, a fraction of stress-induced wild-type p53 rapidly translocates to mitochondria and initiates a first wave of MOMP 3, 4, 5, 6. This seems to be a universal response in p53-mediated apoptosis that occurs in primary and transformed cells, as well as in mice in radiosensitive tissues in response to the gamut of p53-inducing death stimuli like DNA damage, hypoxia and oncogene deregulation 2. Of note, tumor-derived DNA-binding mutants have concomitantly lost their binding to BclXL/Bcl2 and their ability to release cytochrome C (cyto C), indicating that p53 mutations in tumors might be 'double hits' 5, 7. On the other hand, mitochondrially targeted wild-type p53 fusion proteins that bypass the nucleus are sufficient to launch effective apoptosis in p53-null cells 4, 5 and exert tumor suppressor activities in vivo against primary myc-driven lymphomas of p53−/−, ARF−/− or mutant p53 backgrounds 8, 9.
MOMP, believed to depend on the opening of the dynamic Bax/Bak-lipid pore, is triggered by BH3-only proteins 10, 11. MOMP is a prerequisite of apoptosis since it liberates essential apoptogenic factors that are sequestered in the intermembranous space between outer and inner membranes. The Bcl2 superfamily of proteins is the central upstream regulator of MOMP. They fall into three different groups. Members of the BH3-only subfamily are the initiators of a chain of events whose end point is the activation of the ultimate pro-apoptotic BH123 effectors Bax and Bak. BH3-only proteins themselves fall into two subgroups, activators and enablers. Activator BH3-only proteins such as tBid function by transiently binding to and directly activating Bax and Bak by inducing their oligomerization. Enabler BH3-only proteins such as Puma promote MOMP more indirectly. They activate Bax and Bak by forming inhibitory complexes with the anti-apoptotic BH1-4 proteins such as Bcl2, BclXL and Mcl1 that normally stabilize the outer membrane 12. As a consequence, these inhibitory enabler complexes are thought to liberate activator BH3-only proteins or Bax and Bak themselves from the pre-existing embrace by the anti-apoptotic Bcl members to start the cascade. Puma's apoptotic activity is due to its strong binding and neutralization of all members of the anti-apoptotic Bcl2 subfamily 12, 13.
Stress-induced mitochondrial p53 interacts with the anti-apoptotic proteins Bcl2, BclXL and Mcl1 and antagonizes their membrane-stabilizing activity 5, 14. For example, mitochondrial p53 promotes Bak oligomerization by releasing Bak from a pre-existing inhibitory complex with Mcl1 in the outer mitochondrial membrane (OMM) 14. On the other hand, mitochondrial p53 can induce Bak oligomerization and cyto C release by direct physical interaction with Bak itself 14. Thus, mitochondrial p53 functionally resembles a 'super' BH3-only protein, in that it acts both as an enabler and as an activator BH3-only.
Alternatively, a p53 death pathway was proposed that takes place in the cytosol. The model holds that stress-induced cytosolic p53 is initially sequestered into an inactive complex by cytosolic BclXL. In a second phase, which requires p53-mediated transactivation of its target gene Puma by nuclear p53, p53 is liberated from the BclXL complex by Puma, which forms a new Puma/BclXL complex instead. p53 is then free to activate monomeric Bax in the cytosol 15. Thus, the action of cytosolic p53 requires Puma and Bax.
Given the Puma and Bax requirement of the cytosolic p53 action, we therefore set out to determine whether the mitochondrial p53 function equally depends on Puma and Bax. Using isogenic sets of genetically deficient cells or mitochondria isolated from them, we find that the pro-apoptotic signaling of mitochondrial p53 is Puma- and Bax-independent. In addition, we undertook a detailed characterization of the degree of mitochondrial membrane alteration induced by p53 and find it to be severely disruptive.
Results
Death stimulus-induced translocation of p53 to mitochondria is independent of Puma and Bax
Puma and Bax have been implicated in p53's cytosolic action of transcription-independent death. To determine whether Puma and Bax proteins are also required for stress-induced translocation of p53 to mitochondria, we subjected a series of isogenic wild-type p53 harboring HCT116 cell lines that were either proficient or deficient in Puma or Bax to various apoptotic stresses. Subsequently, we determined the level of translocated endogenous p53 in highly purified mitochondria. We chose HCT116 colorectal cancer cells because they are an established model for p53 activation and apoptosis induction in response to multiple stresses 16, 17, 18. Moreover, the transcription-independent mitochondrial p53 pathway was shown to be necessary and sufficient to mediate p53-dependent death in HCT116 cells 19. However, as shown in Figure 1, stress-induced mitochondrial p53 accumulation is Bax- and Puma-independent. In response to DNA strand breaks by camptothecin (CPT), or DNA damage by the nucleotide analog 5′ fluorouracil, or transcriptional blockade by α-amanitin (αAM), which all induce robust p53 stabilization in whole cell lysates, mitochondrial p53 accumulation is completely unaffected by the absence of Puma or Bax (Figure 1A top and bottom).

Death stimulus-induced translocation of p53 to mitochondria is independent of Puma and Bax. (A) HCT116 cells of the indicated genotypes were treated with camptothecin (CPT), α-amanitin (A) or 5′ fluorouracil (F), or left untreated. Whole cell lysates and purified mitochondria (equal total protein) were immunoblotted. Stress-induced p53 translocation to mitochondria is completely unaffected by Puma and Bax deficiency. Mthsp70 is a mitochondrial and PCNA a nuclear contamination marker. (B) p21−/− HCT116 cells or Bax- and Puma-deficient isogenic derivatives were treated with transcriptional inhibitor α-amanitin and characterized as in (A). (C) Blockade of the transcriptional arm of the p53 response does not significantly interfere with caspase 3 activation/PARP cleavage and apoptosis in HCT116 cells. Also, baseline levels of cytosolic Puma promote the mitochondrial arm of p53 apoptosis (see text). Cells of the indicated genotypes were treated with camptothecin (CPT, 5 μM), α-amanitin (A or αAM, 10 μg/ml) or 5′ fluorouracil (F or 5FU, 375 μM) for 24 h (upper panels) or 48 h (lower panels) and immunoblotted as in (A). Apoptosis was assessed by sub-G1 fraction and TUNEL.
These results are confirmed in HCT116 p21−/− cells, which exhibit enhanced apoptotic susceptibility compared to their p21+/+ counterparts due to their impedance in undergoing cell cycle arrest 16, 20. In response to αAM, a highly efficient RNA Pol II inhibitor that induces p53-dependent, but transcription-independent apoptosis in these and other cells 19, 21, total cellular levels of p53 and Ser15-phosphorylated p53 become stabilized. However, their efficient translocation to mitochondria is unimpeded by the absence of Puma or Bax (Figure 1B). These data also indicate that Puma and Bax do not act as mitochondrial anchors for translocated active p53. Moreover, Puma deficiency and αAM-mediated blockade of the transcriptional arm of the p53 response also fail to interfere with several key apoptotic events at mitochondria, such as translocation of monomeric Bax from the cytosol to the outer membrane (Supplementary information, Figure S1) and caspase 3 cleavage/PARP cleavage (Figure 1C, left upper panel, and lower panel). Interestingly, in the first 24 h after DNA damage, αAM-induced transcription-independent apoptosis is of the same magnitude as the apoptosis induced by 5FU, which mediates cell death via both p53 transcription-dependent and -independent pathways. Furthermore, a-amanitin-induced apoptosis in Puma-proficient cells is higher than in Puma-deficient cells (Figure 1C, upper part, see subG1 and TUNEL), indicating some effect of baseline cytosolic Puma present prior to p53-mediated Puma induction. This suggests a Puma-autonomous p53-independent contribution to the death pathway, which has been demonstrated in many p53 null or p53 mutant cell systems 22, 23, 24. Alternatively, baseline Puma might somehow promote the mitochondrial arm of p53 apoptosis via an unknown effect. Prolonged exposure (48 h) to various forms of DNA damage seems to diminish this effect, indicating that indeed pre-induction baseline cytosolic Puma either supports p53 mitochondrial apoptosis at the early onset of cell death or has some p53-independent function that contributes to the execution of the death program during the beginning of the death process but does not have a significant effect when apoptosis is further advanced (Figure 1C, lower panel).
Mitochondrial p53 is highly efficient in inducing MOMP and subsequent release of apoptogenic factors
We previously established that purified p53, when added to isolated unstressed mitochondria, possesses robust release activity for cyto C, a largely intermembranous soluble apoptotic activator, due to p53 protein directly inducing Bak oligomerization and the pivotal process of MOMP 5, 7. Thus, as expected, p53 protein promotes the release of cyto C from purified mitochondria of wild-type HCT116 cells, normal mouse liver and wild-type mouse embryo fibroblasts in a time- and dose-dependent fashion. This action of p53 is similar to that of the prototypical MOMP inducer tBid, an activator BH3-only protein (Figure 2A–2E). Moreover, p53 and tBid have comparable efficiencies in their ability to release other soluble pro-apoptotic factors like Smac 25, 26, also present in the intermembranous space (Figure 2A, 2C, 2E). Beyond this, p53 appears to be able to release the entire gamut of apoptogenic factors, while tBid appears largely restricted to releasing soluble components. For example, apoptosis-inducing factor (AIF) (essential for chromosome condensation) and Endonuclease G, which are both non-soluble factors tethered to the outer surface of the IMM in unstressed mitochondria, are efficiently released by p53 in a dose-dependent fashion (Figure 2A-2C, 2E). It was shown earlier that both MOMP and proteolytic cleavage of mature AIF from its IMM-embedded N-terminus are pre-requisites for AIF release 27, 28. The release of the pro-apoptotic factors cyto C, Smac, AIF and EndoG are dependent on p53's mitochondrial translocation, as shown by concomitant binding of added recombinant p53 to mitochondrial pellets (Figure 2A). Thus, in sum, the p53-induced robust release of AIF and Endo G from mouse and human mitochondria indicates that p53 has a strong permeabilizing effect on mitochondrial membranes that compares favorably with the prototypical activator of the Bax/Bak pore, tBid. Moreover, this suggests that p53 is capable of severely disrupting the integrity of mitochondrial membranes.

Mitochondrial p53 is highly efficient in inducing MOMP and the release of apoptogenic factors and does not require Puma and Bax. Purified mitochondria from (A) HCT116 WT cells or (B) WT mouse liver were incubated with buffer (−), bovine serum albumin (BSA) (400 nM) or increasing amounts of purified p53 (10, 20, 40, 100 nM) or tBid (10, 20, 50, 100 nM). The released apoptogenic factors cyto C, Smac, AIF and Endonuclease G were detected in the supernatants by immunoblotting (equal protein input per lane), and the retained amount of the indicated factors was detected in the pellet fractions. The level of p53 and tBid is shown in supernatants and pellets. S, supernatant; P, mitochondrial pellet. VDAC and CoxIV, mitochondrial loading controls. (C) p53-induced release of cyto C, Smac and AIF is Puma-independent. Mitochondria from WT and PUMA−/− HCT116 cells were incubated with buffer, BSA (100 nM), or increasing amounts of p53 (10, 20, 40 nM) or tBid (10, 20, 50 nM) and immunoblotted as in (A). (D) MOMP activity of p53 is Puma-independent in primary MEFs. Mitochondria from MEFs prepared from WT and PUMA−/− littermates were incubated with purified p53 (10 nM) or tBid (20 nM) and immunoblotted as in Figure 2A. (E) p53-mediated release of cyto C, Smac and AIF is independent of the effector Bax. Analysis as in (A).
Mitochondrial p53-induced MOMP action does not require Puma and Bax
Since p53 translocation is independent of Puma and Bax, we next asked whether the release action of mitochondrial p53 requires Puma or Bax. Low basal levels of Puma and Bax proteins exist in the mitochondria of HCT116 cells in the absence of apoptotic signals 29 (and data not shown). Thus, to assess their requirement for MOMP, we compared the release response of isolated WT, Bax−/− and PUMA−/− mitochondria to purified recombinant p53 and to tBid. Of note, p53 induces the release of cyto C, Smac and AIF in a Puma-independent manner (Figure 2C). Similar results for p53 were obtained with mitochondria from WT and PUMA−/− MEFs and with liver mitochondria from WT and PUMA−/− mice (Figure 2D and data not shown). tBid-mediated release of cyto C and Smac is also Puma-independent (Figure 2C). Moreover, p53-mediated MOMP with subsequent release of cyto C, Smac and AIF is also completely independent of the Bax effector (Figure 2E). In contrast, tBid-mediated release of cyto C and Smac is compromised by Bax deficiency (Figure 2E). The latter result suggests that p53 is flexible with respect to the functionally redundant BH123 effectors Bax and Bak. In the absence of one effector, p53 still mediates a full-range MOMP via the other, while tBid appears to be heavily dependent on Bax.
Mitochondrial p53 mediates oligomerization of Bax and Bak independently of Puma
As we and others have shown previously, the mitochondrial activities of p53 are mediated at least in part via stable protein complexes with resident OMM proteins from the Bcl2 family. Thus, stress-translocated p53 stably interacts with Bcl2, BclXL and Bak via its DNA-binding domain 5, 7, 14. In contrast, a p53-Bax complex has never been observed in the mitochondria, nor anywhere else in the cell. A transient, unstable p53-Bax complex was proposed to occur in the cytosol and mediate Bax activation 15. On the other hand, oligomerization of Bax and Bak in the OMM of mitochondria is the biochemical hallmark of the Bax/Bak lipid pore activation and the process of MOMP. Given the robust MOMP activity of p53 (Figure 2), we therefore assayed the ability of purified p53 to induce Bax and Bak oligomerization using chemical crosslinking. The activator BH3-only protein tBid served again as positive control. Indeed, both p53 and tBid are able to induce Bax-containing oligomers in mitochondria from HCT116 WT cells, but not from their Bax−/− counterparts, indicating the specificity of the oligomeric bands (Figure 3A, panels 1 and 2). Moreover, oligomerization induced by p53 and tBid is also Puma-independent (Figure 3A, panels 1 and 3). Likewise, and as predicted from its MOMP action in Bax−/− cells, p53 also induces oligomerization of Bak in WT, Bax−/− and PUMA−/− mitochondria (Figure 3B). The latter event is associated with formation of an endogenous p53/Bak complex in the mitochondria of stressed wtp53 cells such as ML1 (see Figure 5D) 14. Interestingly, the Bax and Bak oligomerization patterns on isolated mitochondria induced by purified p53 are qualitatively different from the ones induced by purified tBid. p53 strongly promotes low molecular weight complexes containing Bax or Bak and possibly other proteins (Figure 3A and 3B), while tBid promotes high molecular weight species that do not enter the gel (data not shown). In confirmation of the observed in vitro behavior, the p53-associated oligomerization of Bax and Bak can also be captured in vivo. wtp53-harboring ML1 cells are a well-established system for a p53-driven apoptotic response that includes the direct mitochondrial arm. For example, DNA damage of ML1 cells by short-term exposure to CPT is known to induce p53 translocation, interactions with Bcl2 members and apoptosis 4, 30. Chemical crosslinking of mitochondrial lysates from CPT-treated ML1 mitochondria yields multiple oligomeric species of endogenous complexes containing Bak or Bax and probably other proteins, in addition to the monomeric proteins (Figure 3C). These species were either novel (in the case of Bak) or greatly increased in levels (in the case of Bax) when compared to crosslinked untreated ML1 mitochondria. Together, these data indicate that mitochondrial p53 promotes oligomerization and activation of endogenous Bax and Bak in the mitochondria and that this activity does not require Puma.

Mitochondrial p53 mediates oligomerization of Bax and Bak independently of Puma. (A, B) p53 is a potent inducer of Bax and Bak oligomerization. Subsequent to cyto C release with buffer (−), p53 (40 nM) or tBid (100 nM), HCT116 mitochondria (35 μg) were subjected to crosslinking with BMH (10 mM in DMSO for 10 min) or mock-treated (DMSO only). 1/10 of the reaction was immunoblotted for Bax (A) or Bak (B). (A) A specific band containing Bax is detected in WT and PUMA−/− but not in Bax−/− mitochondria. *Non-specific band, also present in Bax−/− cells. (B) A specific band containing Bak is detected in all genotypes. The molecular weight of ∼40 kDa (Bax complex) and ∼50 kDa (Bak complex) includes the possibility of homo-oligomers or hetero-oligomers with a protein X. (C) Death stimulus-induced endogenous mitochondrial p53 translocation correlates with oligomerization of mitochondrial Bak and Bax. wtp53-harboring ML1 cells were mock-treated or treated with CPT (5 μM for 3 h) to induce mitochondrial p53 translocation (right). Purified mitochondria were crosslinked and immunoblotted with N-terminal Bax and Bak antibodies (left). VDAC, mitochondrial loading control.

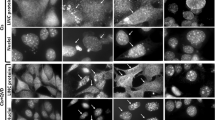
p53 disrupts the integrity of inner and outer mitochondrial membranes. (A) Time-dependent release of mitochondrial matrix-entrapped calcein in cells correlates with stress-induced translocation of endogenous wtp53 to mitochondria. Calcein-labeled ML1 cells were left untreated or treated with CPT (5 μM) for up to 5 h and quenched prior to FACS analysis. Mitochondria exhibit a time-dependent loss of calcein (left), whose kinetics parallels that of CPT-induced mitochondrial translocation of endogenous p53 (right). (B, C) Stress-induced calcein release from the mitochondrial matrix is in part p53-dependent. (B) HCT116 WT and p53 −/− cells were labeled with calcein, stressed with αAM (10 μg/ml), CPT (5 μM), 5FU (375 μg/ml), or ionomycin (500 nM) and quenched prior to FACS analysis. Ionomycin gives the maximum release. (C) Stress-induced calcein release is partially rescued by the p53-specific inhibitor Pifithrin-α (PFT). Calcein-labeled ML1 cells were stressed for 6 h with the hypoximimetic drug desferroxamine (DEX, 125 μM) with or without PFT (20 nM) and quenched prior to FACS analysis. (D) wtp53 forms a DNA damage-induced endogenous complex with Cyclophilin D, a constitutive component of the inner mitochondrial membrane implicated in regulating the PTP pore. Top: Isolated mitochondria from ML1 cells, left untreated or treated with CPT (5 μM for 3 h) to induce mitochondrial p53 translocation, were immunoprecipitated (250 μg total protein each) with the indicated antibodies (1 μg) and blotted for p53. An endogenous stress-induced p53-Bak complex was also detected 14, while p53 did not form a complex with PCNA. Bottom: Corresponding immunoblots of stressed mitochondria from top panel (50 μg each).
Tumor-derived p53 missense mutants have lost the ability to activate the Bax/Bak lipid pore
We showed previously that p53 proteins bearing tumor-derived missense mutations in the DNA-binding domain (DBD) are unable to complex with BclXL and Bcl2 and promote cyto C release 5, 7. We therefore reasoned that the p53 DBD mutants R175H, L194F, R249S and R280K, of which all but the 194 mutant are tumor hotspot mutations, should also be compromised in their ability to alter the oligomerization and functional status of the Bax/Bak lipid pore components in the OMM. This is indeed the case (Figure 4). As expected, wtp53 readily releases cyto C and Smac in a dose-dependent manner (Figure 4A top panels). In contrast, all mutants fail to induce significant release (see 'S' fractions), as seen previously. Importantly, this inability of mutants for MOMP correlates with their pronounced decrease to induce dimeric and higher multimeric Bak and Bax forms (Figure 4A bottom and data not shown). The most severe structural mutant R175H, which is characterized by complete unfolding of the backbone of its DNA-binding domain, is completely disabled to induce Bak dimers, while the milder contact mutants exhibit on average about 50% loss in activity compared to wtp53. Moreover, voltage-dependent anion channel (VDAC, a family of abundant proteins in the OMM) exhibits multimers upon crosslinking in addition to the predominantly monomeric form in unstressed mitochondria. These VDAC multimers disappear with increasing concentrations of wtp53 (Figure 4B, see red dots), suggesting that wtp53 has the ability to drive VDAC into very high molecular weight complexes that no longer enter the gel. In contrast, all p53 mutants have lost the ability to alter the multimeric state of VDAC.

Tumor-derived p53 missense mutants have lost the ability to activate the Bax/Bak lipid pores. (A, B) Purified HCT116 WT mitochondria were incubated with buffer (−), BSA (250 nM) or increasing amounts of purified wild-type or missense mutant p53 proteins (10, 40, 100 nM each), among them the hotspot DNA-binding domain mutants R175H, R249S and R280K. Aliquots were immunoblotted for MOMP (A top, see 'S' fraction) or crosslinked and immunoblotted for Bak (A bottom), Bax (data not shown) and VDAC (B). Blue dot in (A) indicates possible Bak dimer, red dots indicate higher Bak multimers. The immunoblot from A was stripped and reblotted with an antibody specific for VDAC proteins. (B) Red dots in (B) indicate VDAC multimers that are present in unstressed mitochondria and that disappear with increasing concentrations of wild-type but not mutant p53. This suggests that wild-type p53 drives VDAC into high molecular weight complexes that cannot enter the gel.
p53 disrupts the integrity of inner and OMMs
As shown above, purified p53 enables mitochondrial release of the entire gamut of apoptogenic activators, including soluble factors from the intermembranous space (cyto C and Smac) as well as non-soluble factors tethered to the IMM (AIF and EndoG). This suggests that p53 is capable of severely disrupting the integrity of both mitochondrial membranes. To support this conclusion and extend our in vitro observations to cells, we measured the release of calcein from mitochondria, a highly selective fluorescent dye indicator of inner membrane integrity (reviewed in 31). The release of calcein, trapped in the matrix of healthy mitochondria of in vivo labeled cells, requires disruption of both inner and outer membranes. For example, the calcium ionophore ionomycin is severely disrupting both membranes and serves as positive control (Supplementary information, Figure S2A and S2B). In contrast, isolated Bax/Bak pore opening is insufficient for calcein release, as shown by calcein non-responsiveness to tBid (Supplementary information, Figure S2B).
Calcein release from the mitochondrial matrix into the cytoplasm of labeled cells correlates with concomitant mitochondrial translocation of endogenous wtp53 in response to stress (Figure 5A). Calcein-loaded ML1 cells stressed with 5 μM CPT exhibit a time-dependent loss of mitochondrial calcein (Figure 5A left), with the kinetics of parallel CPT-induced translocation of p53 to mitochondria (Figure 5A right). At the highest level of translocation 5 h after the onset of stress, almost 50% of the trapped probe is released (Figure 5A left). To confirm that this effect is p53-dependent and not cell- or stress-type specific, we performed similar studies on the isogenic pair of WT and p53−/− HCT116 cells. Calcein release triggered by αAM, 5FU or CPT is partly p53-dependent, since p53−/− cells show minimal or no response for each drug (Figure 5B). Ionomycin-mediated release, which acts via calcium influx-mediated osmotic rupture of mitochondria, was used as control to obtain the maximum value in this assay. p53-contributed disruption of both inner and outer membranes is further supported by experiments using pharmacological p53 inhibition. Hypoxia-induced calcein release in response to the hypoximimetic drug desferroxamin (DEX) is partially rescued with the p53-specific inhibitor pifithrin-α (Figure 5C). Hypoxia-induced apoptosis very frequently involves the mitochondrial arm of p53-mediated death. While the observed effect of pifithrin-α could be due to inhibition of p53's transcriptional activity, it has been demonstrated that pifithrin-α can also block p53's mitochondrial translocation and subsequent apoptosis in ischemia- and DNA damage-induced death of renal cortex and brain cortex 32, 33, 34. In further support, a stress-inducible endogenous complex between stress-translocated wtp53 and cyclophilin D, a constitutive component of the inner mitochondrial membrane implicated in regulating the PTP pore, is formed in ML1 cells treated with CPT (Figure 5D). This complex is absent in unstressed cells. Taken together, these data suggest that the direct mitochondrial arm of p53 disrupts both the outer and inner mitochondrial membranes in vivo.
Discussion
The direct mitochondrial p53 pathway has been implicated to contribute to p53-driven death in several pathophysiological settings in vivo (reviewed in 35). This includes cell death in radiosensitive healthy tissues such as thymus, spleen and testis after γ-irradiation, and after ischemia/reperfusion injury in rodent models of acute renal failure and hippocampal stroke 36, 37, 38, 39. In the present study, we set out to define the full spectrum of death-promoting activities of mitochondrial p53. We find that p53 and the prototypical activator BH3-only protein tBid are equally competent in activating the Bax/Bak-lipid pore. This in turn enables the release of soluble pro-apoptotic factors from the intermembranous space of mitochondria such as cyto C and Smac, critical for apoptosome activation and IAP inhibition, respectively. In addition, p53 exerts a surprisingly disruptive action on mitochondrial membranes. Thus, p53 effectively liberates the entire gamut of pro-apoptotic factors that include not only the soluble factors that trigger caspase activation but also the insoluble factors such as chromatin condensation and endonuclease activities required for breaking down chromatin and DNA. This inescapably results in cell death. Mitochondrial p53 accomplishes this action by inducing concomitant alterations of the inner and outer membrane. This is reflected by several p53-induced biochemical alterations of membrane components. For example, purified wtp53 protein, but not tumor-derived p53 mutants induce Bax and Bak oligomerization, which are the hallmarks of mitochondria undergoing MOMP. p53 also induces high molecular weight VDAC multimers in the outer membrane. While the precise significance of this remains to be elucidated, it is interesting that VDAC2 was shown to form specific inhibitory complexes with inactive conformers of Bak in unstressed OMMs that prevent Bak oligomerization and mitochondrial apoptosis. Conversely, the BH3-only proteins tBid, Bim or Bad displace VDAC2 from Bak, enabling homo-oligomerization of Bak and apoptosis 40, 41. Moreover, in addition to the known stress-induced complexes of mitochondrial wtp53 with BclXL, Bcl2 and Bak in damaged cells, we found that mitochondrial p53 forms a stress-induced endogenous complex with Cyclophilin D, a component of the inner membrane. Functionally, these changes are associated with marked disruption of inner and outer membrane integrity, which is reflected by the p53-induced release of calcein from the mitochondrial matrix into the cytoplasm of labeled cells. In addition to the mitochondrial p53 action, an alternative cytosolic p53 death pathway was reported that directly activates cytosolic Bax. It occurs in those cell types that express a fraction of BclXL in the cytosol and was described in UV-treated transformed mouse embryonic fibroblasts 15, 42. (The major BclXL fraction resides in the OMM 43, 44. Bcl2 is exclusively membrane-bound and does not appear to participate in this pathway 45.) Here, cytosolic p53 activates monomeric Bax by inducing its homo-oligomerization, followed by Bax mitochondrial translocation. It employs a two-step hybrid mechanism that involves transcription-dependence and -independence. Cytosolic p53, held inactive by a pre-existing soluble p53-BclXL complex, first needs to undergo activation in the following way. Upon stress, nuclear p53 transcriptionally induces the Puma gene. Puma then liberates cytosolic p53 from its BclXL complex by binding to BclXL instead, hence freeing p53 to activate Bax 15, 42. In contrast to the nuclear and mitochondrial p53 functions that are mediated by the p53 DNA-binding domain, here the N-terminus of p53, comprising the first 102 amino acids, and in particular the polyproline-rich domain spanning aa 62-91, is sufficient to mediate Bax activation and apoptosis 42. Importantly, the action of the cytosolic p53 pathway intimately depends on Puma and Bax proteins. Moreover, overexpression experiments with p53 and a BclXL mutant that binds p53 but not Puma in transformed BclXL−/− MEFs showed that ectopic Puma could only reverse the apoptosis-suppressing effect of wild-type but not of mutant BclXL. This latter result implies that binding of p53 to BclXL in the cytosol is by itself not triggering the apoptotic cascade but it requires p53 to be released from BclXL by Puma 15. Furthermore, a recent study demonstrated that disrupting the transcriptional activation of the Puma gene by p53 abrogates DNA damage-induced apoptosis, indicating that transcription-dependent functions of p53 can be necessary in certain scenarios of Puma-mediated apoptosis 46.
In contrast, while Puma is required to couple the nuclear and cytosolic pro-apoptotic function of p53, we show here that mitochondrial p53 does not require Puma, neither for its stress-induced translocation nor for its MOMP function. This result supports the notion that mitochondrial p53, when acting on the outer membrane, mimics a broad-spectrum enabler type BH3-only protein like Puma. Indeed, binding of p53 to Bcl2 was shown to induce a conformational change of Bcl2 that is regulated by its flexible loop, with exposure of Bcl2's BH3 domain and inactivation of its antiapoptotic function 47. Moreover, even in the absence of Bax mitochondrial p53 mediates a full range MOMP activity and releases soluble and insoluble apoptotic activators by signaling via Bak instead. Thus, mitochondrial p53 is flexible with respect to the functionally redundant BH123 effectors Bax and Bak. This appears to be in contrast to the prototypical activator BH3-only protein, tBid, that mainly signals via Bax. Together, our data strengthen the model that mitochondrial p53 functionally resembles a 'super' BH3-only protein, in that it acts both as an enabler and as an activator BH3-only.
Materials and Methods
Cells
The wtp53-harboring human colorectal carcinoma line HCT116 and its isogenic derivatives PUMA−/−, Bax−/−, p21−/−, p21−/− Bax−/−, p21−/−PUMA−/− and p53−/− were provided by B Vogelstein 16. Cells were grown in McCoy's 5A media plus 10% FCS. The human myeloid leukemia line ML1 (wtp53) was grown in RPMI 1640 plus 10% FBS. MEFs from PUMA−/− 48 and WT littermates (E13.5) were prepared by standard protocol and used up to passage 5. αAM, 5′ fluorouracil, CPT and cyclosporine A (Sigma), Ionomycin (Mol. Probes) and BMH (1,6-bis-maleimidohexane, Pierce) were purchased, Pifithrin-α was a gift from A Gudkov.
Mitochondrial isolation and immunoblotting
Mitochondria from mouse liver and cultured cells were isolated by sucrose gradients as described 5, 30. In a few cases they were prepared with the Quick-Prep MitoKit (Pierce). Antibodies were as follows: p53 (DO-1, pSer-15 p53), PCNA, AIF (clone E-1), (all Santa Cruz Biot), mthsp70 and Cyclophilin D (both ABR); COX IV (Molecular Probes); cleaved caspase 3 (#9661) and Bid (both Cell Sign), cleaved PARP (Calbiochem), cyto C (Clone 7H8.2C12, Pharmingen); Smac/Diablo (Clone FKE02, R&D); Endo G (#3035, ProSci); Bax (Ab-1, Oncogene Res and N-20, Santa Cruz); Bak (Bak-NT, Upstate Biotechn), VDAC (pan-human VDAC, Calbiochem). Equal protein from whole cell lysates or subcellular fractions was immunoblotted. CypD and Bak were immunoprecipitated from 500 μg mitochondrial lysate with 1 μg antibody and ProtA Sepharose.
MOMP assays
Release assays for apoptotic activators were performed on mitochondria isolated from freshly harvested mouse liver and cultured cells. Mouse liver mitochondria were gradient-purified 5. Cultured cells were resuspended in mannitol (MB) buffer, swollen for 10 min and dounce-homogenized. Nuclei and unlysed cells were pelleted at 2 000 × g. Mitochondria were recovered from the supernatant by centrifugation at 13 000 × g 49. Mitochondria (350 μg/ml) were incubated with purified p53, tBid (R&D Systems), TypeV BSA (Sigma) or buffer for 20 min at 30 °C and promptly centrifuged at 4 °C 5. Resulting supernatants and washed mitochondrial pellets were analyzed by immunoblotting. His-tagged human p53 proteins were baculovirally (wtp53) or bacterially expressed (wtp53 and p53 DBD mutants R175H, L194F, R249S, R280K and R282W; previously characterized in 7. Protein purity was assessed by Coomassie gels and quantitated by comparing against a BSA standard.
Calcein release assays
Calcein (Molecular Probes) was used to assess mitochondrial membrane integrity in whole cells and isolated mitochondria. Purified mitochondria in MB buffer were labeled for 20 min at 30 °C with 1-10 μM calcein, washed well and incubated with recombinant proteins from 1 to 30 min at 30 °C. Pelleted mitochondria were immediately analyzed by FACS. The corresponding supernatants were subject to immunoblots for cyto C and AIF release. For in situ release, cells were labeled with calcein for 30 min at 37 °C in Hanks' balanced salt solution with 10 mM Hepes, pH 7.4 and 0.1 μM calcein. Following drug treatment or microinjection of purified proteins, cytosolic calcein fluorescence was quenched for 20 min at 37 °C with 1 mM CoCl2. Fluorescence acquisition on whole cells was performed according to the manufacturer's instructions on a FACS Calibur and analyzed by ModFit LT software. Retention of the initial mitochondrial fluorescence intensities is reported after normalization for background (unlabeled). Microinjected cells were analyzed using a Zeiss epifluorescent Axioskope.
Apoptosis analysis
Cells were fixed with 4% paraformaldehyde, permeabilized in 0.5% Tween and processed using the TUNEL Detection Kit TMR Red (Roche). Nuclei were counterstained with 1 mg/ml Hoechst 33342 and slides were counted on a Zeiss epifluorescent Axioskope. Apoptosis was quantified as the proportion of red/blue areas (TUNEL/Hoechst) from 3 representative images, using the Automatic Measurement Program of Zeiss AxioVision v4.3. Alternatively, subG1 was determined with propidium iodide labeled cells via FACS and ModFit LT software.
(Supplementary information is linked to the online version of the paper on the Cell Research website.)
References
Perfettini JL, Kroemer RT, Kroemer G . Fatal liaisons of p53 with Bax and Bak. Nat Cell Biol 2004; 6:386–388.
Moll UM, Marchenko N, Zhang XK . p53 and Nur77/TR3 – transcription factors that directly target mitochondria for cell death induction. Oncogene 2006; 25:4725–4743.
Erster S, Mihara M, Kim RH, Petrenko O, Moll UM . In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004; 24:6728–6741.
Marchenko ND, Zaika A, Moll UM . Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 2000; 275:16202–16212.
Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577–590.
Sansome C, Zaika A, Marchenko ND, Moll UM . Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett 2001; 488:110–115.
Tomita Y, Marchenko N, Erster S, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem 2006; 281:8600–8606.
Talos F, Petrenko O, Mena P, Moll UM . Mitochondrially targeted p53 has tumor suppressor activities in vivo. Cancer Res 2005; 65:9971–9981.
Palacios G, Moll UM . Mitochondrially targeted wild-type p53 suppresses growth of mutant p53 lymphomas in vivo. Oncogene 2006; 25:6133–6139.
Kuwana T, Mackey MR, Perkins G, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002; 111:331–342.
Chipuk JE, Green DR . Dissecting p53-dependent apoptosis. Cell Death Differ 2006; 13:994–1002.
Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 2005; 17:525–535.
Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17:393–403.
Leu JI, Dumont P, Hafey M, Murphy ME, George DL . Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443–450.
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR . PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005; 309:1732–1735.
Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L . PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 2003; 100:1931–1936.
Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B . Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev 1996; 10:1945–1952.
Kaeser MD, Pebernard S, Iggo RD . Regulation of p53 stability and function in HCT116 colon cancer cells. J Biol Chem 2004; 279:7598–7605.
Arima Y, Nitta M, Kuninaka S, et al. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem 2005; 280:19166–19176.
Waldman T, Kinzler KW, Vogelstein B . p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187–5190.
Derheimer FA, Chang CW, Ljungman M . Transcription inhibition: a potential strategy for cancer therapeutics. Eur J Cancer 2005; 41:2569–2576.
Callus BA, Ekert PG, Heraud JE, et al. Cytoplasmic p53 is not required for PUMA-induced apoptosis. Cell Death Differ 2008; 15:213–215.
Nakano K, Vousden KH . PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683–694.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673–682.
Du C, Fang M, Li Y, Li L, Wang X . Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102:33–42.
Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102:43–53.
Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ . Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 2003; 22:4385–4399.
Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG . Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem 2005; 280:6447–6454.
Yamaguchi H, Chen J, Bhalla K, Wang HG . Regulation of Bax activation and apoptotic response to microtubule-damaging agents by p53 transcription-dependent and -independent pathways. J Biol Chem 2004; 279:39431–39437.
Mihara M, Moll UM . Detection of mitochondrial localization of p53. Methods Mol Biol 2003; 234:203–209.
Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F . The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem 2001; 276:12030–12034.
Gilman CP, Chan SL, Guo Z, Zhu X, Greig N, Mattson MP . p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromol Med 2003; 3:159–172.
Kelly KJ, Plotkin Z, Vulgamott SL, Dagher PC . P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol 2003; 14:128–138.
Strom E, Sathe S, Komarov PG, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2006; 2:474–479.
Erster S, Moll UM . Stress-induced p53 runs a direct mitochondrial death program: its role in physiologic and pathophysiologic stress responses in vivo. Cell Cycle 2004; 3:1492–1495.
Bonini P, Cicconi S, Cardinale A, et al. Oxidative stress induces p53-mediated apoptosis in glia: p53 transcription-independent way to die. J Neurosci Res 2004; 75:83–95.
Dagher PC . Apoptosis in ischemic renal injury: roles of GTP depletion and p53. Kidney Int 2004; 66:506–509.
Endo H, Kamada H, Nito C, Nishi T, Chan PH . Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J Neurosci 2006; 26:7974–7983.
Endo H, Saito A, Chan PH . Mitochondrial translocation of p53 underlies the selective death of hippocampal CA1 neurons after global cerebral ischaemia. Biochem Soc Trans 2006; 34:1283–1286.
Chandra D, Choy G, Daniel PT, Tang DG . Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J Biol Chem 2005; 280:19051–19061.
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ . VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003; 301:513–517.
Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010–1014.
Gonzalez-Garcia M, Pérez-Ballestero R, Ding L, et al. bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development 1994; 120:3033–3042.
Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C . Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 2003; 160:53–64.
Hsu YT, Wolter KG, Youle RJ . Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 1997; 94:3668–3672.
Wang P, Yu J, Zhang L . The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci USA 2007; 104:4054–4059.
Deng X, Gao F, Flagg T, Anderson J, May WS . Bcl2's flexible loop domain regulates p53 binding and survival. Mol Cell Biol 2006; 26:4421–4434.
Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003; 4:321–328.
Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC . Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA 1998; 95:4997–5002.
Acknowledgements
This work was supported by grants to UMM from the National Cancer Institute (USA) and from the Carol M. Baldwin Breast Cancer Award.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary information, Figure S1
Death stimulus-induced mitochondrial translocation of Bax is independent of Puma. (PDF 155 kb)
Supplementary information, Figure S2
HCT116 WT cells were loaded in situ with the fluorescent dye calcein that gets trapped in the mitochondrial matrix. (PDF 136 kb)
Rights and permissions
About this article
Cite this article
Wolff, S., Erster, S., Palacios, G. et al. p53's mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res 18, 733–744 (2008). https://doi.org/10.1038/cr.2008.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2008.62
Keywords
This article is cited by
-
Targeting the overexpressed mitochondrial protein VDAC1 in a mouse model of Alzheimer’s disease protects against mitochondrial dysfunction and mitigates brain pathology
Translational Neurodegeneration (2022)
-
Mutant p53 in cancer: from molecular mechanism to therapeutic modulation
Cell Death & Disease (2022)
-
Targeting Apoptosis in Cancer
Current Oncology Reports (2022)
-
Resveratrol as a modulatory of apoptosis and autophagy in cancer therapy
Clinical and Translational Oncology (2022)
-
TMEM166 inhibits cell proliferation, migration and invasion in hepatocellular carcinoma via upregulating TP53
Molecular and Cellular Biochemistry (2021)
