Actinide chemistry
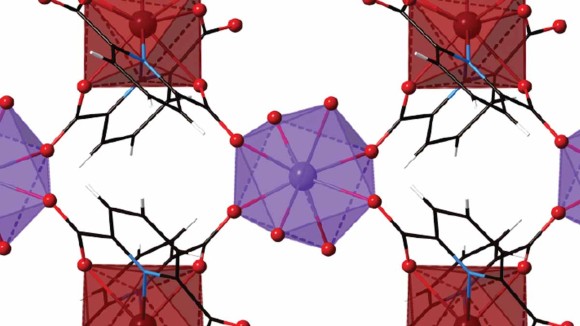
Our understanding of the bonding, reactivity and electronic structure of actinides, though it has both fundamental and practical importance, lags behind that of the rest of the periodic table. A collection of articles in this Focus highlights recent developments in this area, in particular featuring uranium(VI) dianions bearing four U–N multiple bonds, berkelium(IV) stabilized in aqueous solution and a plutonium material showing evidence for the delocalization of 5f electrons.