
Abstract
Skin wound macrophages are key regulators of skin repair and their dysfunction causes chronic, non-healing skin wounds. Peroxisome proliferator-activated receptor gamma (PPARγ) regulates pleiotropic functions of macrophages, but its contribution in skin wound healing is poorly defined. We observed that macrophage PPARγ expression was upregulated during skin wound healing. Furthermore, macrophage PPARγ deficiency (PPARγ-knock out (KO)) mice exhibited impaired skin wound healing with reduced collagen deposition, angiogenesis and granulation formation. The tumor necrosis factor alpha (TNF-α) expression in wounds of PPARγ-KO mice was significantly increased and local restoration of TNF-α reversed the healing deficit in PPARγ-KO mice. Wound macrophages produced higher levels of TNF-α in PPARγ-KO mice compared with control. In vitro, the higher production of TNF-α by PPARγ-KO macrophages was associated with impaired apoptotic cell clearance. Correspondingly, increased apoptotic cell accumulation was found in skin wound of PPARγ-KO mice. Mechanically, peritoneal and skin wound macrophages expressed lower levels of various phagocytosis-related molecules. In addition, PPARγ agonist accelerated wound healing and reduced local TNF-α expression and wound apoptotic cells accumulation in wild type but not PPARγ-KO mice. Therefore, PPARγ has a pivotal role in controlling wound macrophage clearance of apoptotic cells to ensure efficient skin wound healing, suggesting a potential new therapeutic target for skin wound healing.
Similar content being viewed by others

Main
The skin is the largest human organ that is essential to protect body against infection and excessive water loss. However, skin is also very easily injured by various attacks.1 After injury, the skin needs to restore homeostasis, structure integrity and functional competence.2 Skin wound healing is a complicated process orchestrated by interactions of inflammatory cells, resident cells, extracellular matrix components and soluble mediators. The healing process is usually divided into three sequential and overlapping phases: inflammation, proliferation and maturation.3 The inflammatory phase includes platelet aggregation, blood coagulation and inflammatory cells recruitment to wound sites. The proliferative phase involves keratinocytes, fibroblasts and endothelial cells migration and proliferation, contributing to reepithelialization, collagen deposition and angiogenesis. And the maturation phase restores tissue structure integrity and functional competence.2 If wounds do not progress in the timely and orderly manner, they convert into chronic, non-healing wounds that are a growing world health-care problem related with increasing incidence of diabetes, obesity and aging.4, 5, 6
Macrophages are the most important immune cells recruited to the wound sites following skin injury, which exhibit pleiotropic functions to orchestrate the healing process throughout the different phases.1, 7, 8 During the earlier inflammation phase, macrophages characterize an pro-inflammatory phenotype, they release pro-inflammatory mediators such as tumor necrosis factor alpha (TNF-α), nitric oxide and IL-6, and produce protease and reactive oxygen species to combat contaminating organisms. In pathogen spread wounds, macrophages phagocytose these pathogens and present antigen to T cells. Macrophages also phagocytose wound debris and apoptotic cells.9 Phagocytosis of apoptotic cells predominately switches pro-inflammatory macrophages to anti-inflammatory/wound-healing macrophages to resolve wound inflammation and initiate the healing process.10, 11 During the later healing phase, macrophages characterize anti-inflammatory/wound-healing phenotype, they produce many cytokines, chemokines and growth factors to crosstalk with keratinocytes, fibroblasts and endothelial cells, contributing to reepithelialization, collagen deposition, angiogenesis, granulation tissue formation and wound repair.12 In wound healing, non-functional or dysfunctional macrophages are related with chronic, non-healing wounds.7, 13, 14, 15 Therefore, sustaining macrophage normal functions is critical for successful wound healing. However, the underlying factors regulate macrophage functions during wound healing are not fully known.
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear receptor superfamily, which includes three isotypes, PPARα, PPARβ/δ and PPARγ. In skin wound healing, PPARα participates in the control of the early inflammation phase of the healing,16 PPARβ regulates keratinocytes proliferation, adhesion and migration;16, 17, 18 and PPARδ promotes fibroblast proliferation.19 However, the role of PPARγ in wound healing is not elucidated. It is well known that PPARγ is a key factor transcriptionally coordinates macrophage functions.20 Macrophage PPARγ signaling is essential for the efficient clearance of apoptotic cells21, 22 and the switching from pro-inflammatory macrophages to anti-inflammatory macrophages,23, 24 which are important for resolving inflammation and maintaining homeostasis. In this study, we generated mice with macrophage PPARγ deficiency to investigate the role of macrophage PPARγ in the healing of skin wounds.
Results
PPARγ is upexpressed in wounded skin and wound macrophage
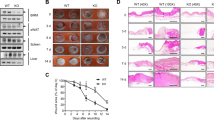
We first investigated the temporal and spatial expression of PPARγ during skin wound healing in wild-type (WT) mice (Figures 1a and b). Low levels of PPARγ (mRNA and protein) were observed in unwounded control skin (day 0). However, a significant increase of mRNA and protein levels of PPARγ was observed after wounding (days 3, 5, 7, 10 and 12). Immunohistochemical staining showed that PPARγ protein was significantly enhanced in both subcutaneous (s.c.) and dermis of wounded skin (Figure 1c) compared with normal skin (Supplementary Figure 1). In addition, flow cytometric analysis showed that wound macrophage upregulated PPARγ expression during the healing process (Figure 1d). These results suggest a potential involvement of macrophage PPARγ in the regulation of skin wound healing.

PPARγ expression during wound healing of WT mice. (a) mRNA and (b) protein levels of PPARγ in wounds. mRNA expression (a) is normalized to β-actin and the protein expression (b) is normalized to GAPDH. *P<0.05, wounded skin versus normal skin. (c) Immunohistochemical staining of PPARγ expression in wounded skin on day 5 after wounding. Boxed areas of s.c. (number 1) and dermis (number 2) tissue in the left panel are enlarged in the middle and right panel. Black hatched line lines eschar. E, eschar. Scale bar=50 μm. (d) Flow cytometric analysis of PPARγ expression in wound macrophages on days 3, 5 and 7. CD11b+F4/80+ was used to gate macrophages. Isotype control, gray histogram; PPARγ, unshaded histogram. Mean fluorescence intensity (MFI) is shown. *P<0.05. Data are expressed as mean±S.D. and images are representative, n=3 for each time point
Characterization of macrophage PPARγ deficiency mice
To investigate the role of macrophage PPARγ in wound healing, conditional knock out (KO) mice lacking macrophage expression of PPARγ were generated by crossing mice bearing the lox-P-targeted PPARγ (PPARγf/f) allele with mice bearing the lysozyme-M Cre (LysMCre) recombinase transgene. We refer to PPARγf/fLysMCre− mice as control (PPARγ-WT), and their PPARγf/fLysMCre+ littermates as KO animals (PPARγ-KO).
Genotyping of the tail DNA confirmed the presence of Cre transgene in PPARγf/+LysMCre+ and PPARγf/fLysMCre+ mice and its absence in WT mice (Figure 2a). Peritoneal macrophages from PPARγ-KO mice showed significant lower levels of PPARγ mRNA and protein compared with their control macrophages (Figures 2b and c), and wound macrophages from PPARγ-KO mice had lower PPARγ expression (Figure 2d). In addition, both PPARγ-WT and PPARγ-KO wound neutrophils showed no evidence for PPARγ staining, and PPARγ expression in splenic T cells, B cells and dendritic cells were not significantly different between PPARγ-WT and PPARγ-KO mice (Supplementary Figure 2). These results indicated an efficient and specific macrophage PPARγ ablation.

Characterization of macrophage-specific PPARγ deficiency mice. (a) Genotyping analysis of PPARγf/fLysMCre+, PPARγf/+LysMCre+ and PPARγ+/+LysMCre− (PPARγ-WT) mice. (b) WB and (c) RT-PCR analysis of PPARγ expression in isolated peritoneal PPARγ-WT and PPARγ-KO macrophages. Protein expression is normalized by GAPDH. mRNA expression is normalized by β-actin and represented as the fold change in PPARγ-KO macrophages compared with PPARγ-WT macrophages. (d) Flow cytometric analysis of PPARγ expression in isolated PPARγ-WT and PPARγ-KO wound macrophages. CD11b+F4/80+ was used to gate macrophages. Isotype control, gray histogram; PPARγ, unshaded histogram (red histogram: PPARγ-WT; black histogram: PPARγ-KO). Mean fluorescence intensity (MFI) is shown. For (b–d), *P<0.05, PPARγ-KO versus PPARγ-WT. (e) mRNA and protein levels of PPARγ in wounds of WT and PPARγ-KO mice. mRNA expression is normalized by β-actin and protein expression is normalized by GAPDH. *P<0.05. Data are expressed as mean±S.D. and images are representative, n=3 for each time point and group
In addition, the temporal profile of PPARγ mRNA and protein levels in skin wounds were compared between PPARγ-KO and PPARγ-WT mice. Significant lower levels of PPARγ (mRNA and protein) were observed in PPARγ-KO mice compared with WT mice after wounding (days 3, 5, 7 and 10; Figure 2e), indicating an important contribution of macrophage PPARγ to the increased PPARγ expression observed during skin wound healing.
Delayed wound healing in mice with macrophage PPARγ deficiency
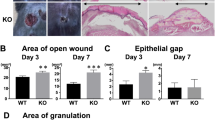
Thereafter, full-thickness circular wounds were produced on PPARγ-WT and PPARγ-KO mice. The wound sizes were monitored daily. In addition, we found that wound closure in PPARγ-KO mice was significantly delayed from 3 to 12 after wounding compared with PPARγ-WT mice (Figure 3a). Next, we detected granulation formation, collagen deposition and angiogenesis during wound healing, focusing on days 5 and 7, when mainly granulation tissue formation, collagen deposition and angiogenesis occur.

Delayed wound healing in PPARγ-KO mice. (a) Representative wounds, and statistical analysis of wound areas expressed as percentage of the initial (day 0) wound size. n=12, for each time point and group. (b) The areas of granulation tissue in 5-day-old wounds. (c) mRNA expression of collagen type 1 in wounds. (d) Quantification of new blood vessels in the granulation tissue of 5-day-old wounds in 3–5 high-power fields (HPFs) per mice, and mRNA expression of VEGF in wounds. For (b–d), n=3 for each time point and group. All data are expressed as mean±S.D. *P<0.05, PPARγ-WT versus PPARγ-KO mice
Hematoxylin and eosin (H&E) staining of 5-day-old wounds showed that areas of granulation tissue was markedly reduced at the wound sites in PPARγ-KO mice compared with PPARγ-WT mice (Figure 3b and Supplementary Figure 3a).
To examine collagen deposition, 5- and 7-day-old wounds were stained with Masson trichrome. At the wound edge, characterized by the acanthotic keratinocyte layer, new and old collagen can be observed. The old collagen away from the wound margin in both strains of mice stains dark blue (Supplementary Figure 3b, closed arrow). The newly formed collagen near the wound margin is below the acanthotic keratinocyte layer (Supplementary Figure 3b, open arrow). Both 5- and 7-day-old wounds in PPARγ-KO mice contained lower areas of new collagen compared with the PPARγ-WT wounds (Supplementary Figure 3b). To further quantitatively assess collagen deposition, collagen type 1 mRNA expression was analyzed by real-time polymerase chain reaction (RT-PCR) and a significantly higher level was detected on days 3, 5 and 7 wounds of PPARγ-WT mice compared with that of PPARγ-KO mice (Figure 3c).
To examine the rate of angiogenesis, immunohistochemistry analysis of CD31 was carried out. The wounds of PPARγ-WT had higher density of new blood vessels in the granulation tissue compared with wounds of PPARγ-KO mice (Figure 3d and Supplementary Figure 3c). Consistently, the mRNA level of vascular endothelial growth factor (VEGF) in wounds was significantly decreased in PPARγ-KO mice compared with PPARγ-WT mice (Figure 3d).
Overall, wound healing is severely impaired both macroscopically and microscopically in PPARγ-KO mice.
Wounds of PPARγ-KO mice exhibit normal numbers of neutrophils and macrophages
It is well known that inflammatory cells have a vital role in normal skin wound healing and impairing inflammatory cell recruitment to wound sites severely affects wound healing.25, 26, 27, 28, 29, 30, 31 So we next measured the leukocyte accumulation to skin wound sites between PPARγ-WT and PPARγ-KO mice. Immunohistochemistry staining of day 3 wounds with antibodies specific to neutrophils (Ly-6G) or macrophages (F4/80) were shown (Supplementary Figure 4). The numbers of neutrophils and macrophages were counted within the granulation tissues of wounds at indicated time points and no significant difference between both mice strains was seen (Figure 4a). Furthermore, flow cytometry was used to measure the numbers of neutrophils and macrophages in skin wound on days 1, 2 and 3 after polyvinyl alcohol (PVA) sponge insertion, and no significant differences were observed between both mice strains as well (Figure 4b). These results indicated that the recruitment of neutrophils and macrophages to skin wound in PPARγ-WT and PPARγ-KO mice are similar.

There is no difference in inflammatory cell recruitment to the wounds of PPARγ-WT and PPARγ-KO mice. (a) Quantification of Ly-6G+ neutrophils within granulation tissues of 1- and 3-day-old wounds, and F4/80+ macrophages within granulation tissues in 3- and 5-day-old wounds in 3–5 high-power fields (HPFs) per mice. (b) Wound cells derived from PVA sponges after days 1, 2 and 3 inserting. The numbers of neutrophils and macrophages were shown. SSChighLy-6G+ was used to gate neutrophils and FSChighF4/80+ was used to gate macrophages. Data are expressed as mean±S.D., n=3 for each time point and group
Local restoration of TNF-α rescues impaired wound healing in PPARγ-KO mice
Other than inflammatory cells, cytokines, growth factors and chemokines regulate the wound healing as well. Accumulated evidences indicate that enhancing local TNF-α expression delays wound healing, reduces collagen deposition and suppresses angiogenesis.32, 33, 34, 35, 36, 37 So we next measured TNF-α levels in wound tissues of PPARγ-KO versus PPARγ-WT mice by RT-PCR, western blotting (WB) and ELISA. Neither mRNA levels nor protein expression of TNF-α have significant difference between PPARγ-KO and PPARγ-WT mice in day 0 wounds (Figures 5a and b). After injury, the expression levels of TNF-α were increased in 5-day-old wounds of both strains mice. However, a significant higher expression of TNF-α both at mRNA and protein level were observed in 5-day-old wounds of PPARγ-KO mice compared with PPARγ-WT mice (Figures 5a and b).

Enhanced expression of TNF-α in wounds is causal for the wound healing defect of PPARγ-KO mice. (a) mRNA and (b) protein expression of TNF-α in normal skin and 5-day-old wounds. (c) Representative wounds, and statistical analysis of wound areas expressed as percentage of the initial (day 0) wound size. n=12, for each time point and group. (d) The areas of granulation tissue in 5-day-old wounds. (e) mRNA expression of collagen type 1 in wounds. (f) Quantification of new blood vessels in the granulation tissue of 5-day-old wounds in 3–5 high-power fields (HPFs) per mice, and mRNA expression of VEGF in wounds. For (a), (b), (d), (e) and (f), n=3 for each time point and group. All data are expressed as mean±S.D. and images are representative. *P<0.05
These results suggested a significant increase of TNF-α following macrophage PPARγ deficiency, which may account for the delayed wound healing. So we next s.c. injected rat anti-mouse TNF-α (aTNF-α) around the wound.35, 36 In addition, mock injections of isotype control antibody were taken as controls. In PPARγ-KO mice, local injection of aTNF-α improved skin wound healing, granulation tissue formation, collagen deposition and angiogenesis to the level similar to that in WT mice without any treatment, indicating a full rescue (Figures 5c–f and Supplementary Figures 5a and c). These data suggest that the increased local TNF-α expression is causal to the delayed wound healing in PPARγ-KO mice.
Impaired apoptotic cell clearance by macrophages contributes to increased wound TNF-α expression in PPARγ-KO mice
As macrophages represent a major source of the cytokines in wounds,38 we speculated that PPARγ-KO macrophages may release more TNF-α compared with PPARγ-WT macrophages during wound healing. Therefore, we detected TNF-α expression in isolated wound macrophages and found that PPARγ-KO wound macrophages expressed more TNF-α than their PPARγ-WT counterparts (Figure 6a).

Increased production of TNF-α is due to impaired macrophage phagocytosis in PPARγ-KO mice. (a) Intracellular staining for TNF-α in wound macrophages derived from PVA sponges after 5 days implantation. F4/80+ was used to gate macrophages. Isotype control, gray histogram; TNF-α, unshaded histogram (red histogram: PPARγ-WT; black histogram: PPARγ-KO). Mean fluorescence intensity (MFI) is shown. The mRNA (b) and protein (c) levels of TNF-α in PPARγ-WT and PPARγ-KO peritoneal macrophages, and in ATs. *P<0.05. #P<0.05, ATs versus PPARγ-WT or PPARγ-KO macrophages stimulated by LPS. (d) Representative flow cytometric raw data of the phagocytosis assays (macrophages were stained using F4/80 PE Ab; ATs were loaded with CFSE), and the percentages of macrophages ingesting ATs. (e) Quantification of TUNEL+ cells in 3-, 5- and 7-day-old wounds in 3–5 high-power fields (HPFs) per mice. (f) The percentages of apoptotic neutrophils in total wound cells derived from PVA sponges after day 3 inserting. Data are expressed as mean±S.D. and images are representative, n=3 for each time point and group. For (a), (d), (e) and (f), *P<0.05, PPARγ-WT versus PPARγ-KO mice
To explore how PPARγ regulates wound macrophage TNF-α production, we next analyzed TNF-α mRNA levels in peritoneal macrophages of both mice strains stimulated with or without lipopolysaccharide (LPS). Unstimulated PPARγ-WT and PPARγ-KO macrophages expressed similar low-level of TNF-α (Figure 6b). Although LPS greatly upregulated TNF-α expression in PPARγ-WT and PPARγ-KO macrophages, no significant difference was observed between these two stains of macrophages (Figure 6b). In addition, similar results were further confirmed at protein level (Figure 6c), indicating that PPARγ has no direct effect on TNF-α production in macrophages after LPS stimulation. However, in the presence of apoptotic thymocytes (ATs), PPARγ-WT macrophages significantly reduced LPS-induced TNF-α production (at both mRNA and protein level), but PPARγ-KO macrophages were not (Figures 6b and c), suggesting that the relative increased TNF-α expression in PPARγ-KO macrophages was because of the failure of ATs phagocytosis. For further demonstration, the actin-filament polymerization-blocking agent cytochalasin B was used to inhibit macrophage phagocytic activity and the added ATs were found to have no effect on TNF-α expression in both macrophages strains (Figures 6b and c). Furthermore, the direct measurement of ATs engulfment by flow cytometry clearly showed that the phagocytic activity of PPARγ-KO macrophage was severely impaired (Figure 6d). These in vitro results suggested that PPARγ-KO macrophages produced excessive TNF-α because of an impaired phagocytic activity.
For in vivo study, TUNEL staining was applied to detect apoptotic cell accumulation in skin wound. We observed that the wounds (days 3, 5 and 7) of PPARγ-KO mice contained higher number of apoptotic cells compared with that in PPARγ-WT mice (Figure 6e and Supplementary Figure 6a). Furthermore, wound cells of PPARγ-KO mice had higher percentage of apoptotic neutrophils compared with PPARγ-WT mice (Figure 6f), indicating decreased apoptotic cell engulfment by PPARγ-KO macrophages in skin wound.
Shortly, these results indicated that PPARγ deficiency impaired macrophage phagocytosis of apoptotic cells, resulting in macrophage producing of excessive TNF-α, contributing to increased TNF-α level in wounds.
PPARγ regulates the expression of genes involved in the phagocytosis of apoptotic cells by macrophages
To understand the underlying molecular mechanism of the deficit in apoptotic cell clearance observed in our mouse model, we profiled the transcription of a set of genes encoding phagocytosis-associated receptors and opsonins in PPARγ-WT and PPARγ-KO macrophages. Among them, Cd36 and Mertk are cell surface receptors and required for proper binding and consequent internalization of apoptotic cells.39 Mfge8, Gas6, and the C1q subunits C1qa, C1qb and C1qc are opsonins, which bind to apoptotic cell surface to initiate phagocytosis.40
In PPARγ-WT and PPARγ-KO macrophages, the expression of Cd36, Mertk, Mfge8, C1qb and C1qc was reduced in PPARγ-KO macrophages. However, PPARγ deficiency had no effect on the expression of Gas6 and C1qa (Figure 7a). As digestion of apoptotic cells by macrophage leading to accumulation of cellular components, such as cholesterol and fatty acids, which may act as endogenous ligands for PPARγ,41 we incubated both strains of macrophage with ATs, the results showed the mRNA levels of Cd36, Mertk, Mfge8, Gas6 and all three C1q subunits were higher in PPARγ-WT macrophages compared with the absence of ATs, which was not observed in PPARγ-KO macrophages (Figure 7a). Furthermore, activation of PPARγ using rosiglitazone (RSG), which is a selective agonist of PPARγ, increased the expression of Cd36 and Mertk in PPARγ-WT macrophages but not in PPARγ-KO cells. However, RSG did not significantly increase the expression of Mfge8, Gas6, C1qa, C1qb and C1qc in PPARγ-WT macrophages (Figure 7b). In addition, isolated macrophages from wound of PPARγ-KO mice decreased the expression of all the detected phagocytosis-related genes (Figure 7c). These results indicated that PPARγ deficiency in macrophages decreased the expression of phagocytosis-related molecules, which may contribute to the reduced apoptotic cell clearance.

PPARγ regulate the expression of macrophages receptors and opsonins involved in the engulfment of apoptotic cells. (a) mRNA expression of phagocytosis-related genes in peritoneal macrophages not added ATs or added ATs. (b) mRNA expression of phagocytosis-related genes in peritoneal macrophages treated with vehicle (ethanol) or RSG. (c) mRNA expression of phagocytosis-related genes in wound macrophages derived from PVA sponges after 5 days implantation. *P<0.05. #P<0.05. αP<0.05, PPARγ-KO macrophages versus PPARγ-WT macrophages. mRNA expression is normalized by β-actin, and gene expression is represented as fold change compared with PPARγ-WT macrophages, or PPARγ-WT or PPARγ-KO macrophages treated with Vehicle. Results are expressed as mean±S.D., n=3 for each group
Therapeutic targeting PPARγ accelerates skin wound healing in normal mice
Owing to PPARγ upexpression in wounded skin and delayed wound healing in mice with macrophage PPARγ deficiency, we speculated that PPARγ may have a vital role in regulating skin wound healing. RSG treatment of skin wound in WT mice resulted in accelerated wound healing, reduced apoptotic cell aggregation and lower local TNF-α expression compared with a control treatment (Figures 8a–c and Supplementary Figure 6b). However, RSG has no therapeutic effects in PPARγ-KO mice (Figures 8a–c and Supplementary Figure 6b).

PPARγ agonist accelerates wound healing in WT mice and suppresses apoptotic cell aggregation and TNF-α production in wounds, but not in PPARγ-KO mice. (a) Representative wounds, and statistical analysis of wound areas expressed as percentage of the initial (day 0) wound size. n=12, for each time point and group. *P<0.05, RSG-treated WT mice versus vehicle-treated WT mice; #P<0.05, RSG-treated PPARγ-KO mice versus vehicle-treated WT mice. (b) Quantification of TUNEL+ cells in 5-day-old wounds in 3–5 high-power fields (HPFs) per mice. (c) mRNA expression of TNF-α in normal skin and 5-day-old wounds. Both (b and c), n=3 for each time point and group. All data are expressed as mean±S.D. *P<0.05. (d) After tissue injury, wound macrophage PPARγ is upregulated to timely disposal of apoptotic cells, resulting in decreased local TNF-α expression to enhancing wound healing
Discussion
PPARγ has a key role in regulating macrophage phagocytic activity during skin wound healing. In this study, PPARγ was upregulated in wounded skin and wound macrophages indicating that macrophage PPARγ may regulate the healing process. Thus, we generated macrophage PPARγ deficiency mice. These mice exhibited efficient and specific macrophages PPARγ ablation, not targeting other immune cells, such as neutrophils, dendritic cells, T cells and B cells, which may influence the skin wound inflammation. Furthermore, the expression of PPARγ in skin was not upregulated during wound healing in PPARγ-KO mice, suggesting an ablation of PPARγ in wound macrophage. PPARγ-KO mice reduced granulation tissue formation, collagen deposition and angiogenesis, and delayed wound closure. In PPARγ-KO mice, the TNF-α expression was increased in skin wound and local inhibition of TNF-α rescued healing deficit in PPARγ-KO mice, indicating that the excessive TNF-α contributed to the impaired skin wound healing in PPARγ-KO mice. Next, we observed that PPARγ-KO wound macrophages released more TNF-α compared with their control macrophages. Furthermore, PPARγ deficiency impaired macrophage phagocytosis of apoptotic cells through downregulating phagocytosis-related molecules in macrophage, resulting in excessive TNF-α expression by PPARγ-KO macrophages. Meanwhile, increased apoptotic cells accumulation in PPARγ-KO wounds was observed. These results directly or indirectly demonstrated that the impaired macrophage phagocytosis contributes to the excessive TNF-α in wound of PPARγ-KO mice. Therefore, macrophage PPARγ deficiency decreased the expression of phagocytosis-related molecules, leading to deficit in clearance of apoptotic cells, resulting in impaired skin wound healing.
The timely and efficient clearance of apoptotic cells by macrophages is the key to skin wound healing.11 Macrophage phagocytosis of apoptotic cells prevents further tissue damage by protecting tissue from exposure to toxic components of dying cells, and suppresses the production of pro-inflammatory cytokines, and enhancing the expression of anti-inflammatory cytokines and growth factors, resulting in resolution of wound inflammation and promoting wound healing.42, 43 Deficit in macrophage phagocytosis function during wound healing are related with chronic, non-healing wounds.13, 14, 15 Our study provides the first evidence of PPARγ regulating wound macrophage phagocytosis function during skin wound healing. In addition, PPARγ deficiency resulted in impaired macrophage phagocytosis function, contributing to delayed wound closure, which is consistent with the importance of apoptotic cell clearance in wound healing. Although several studies have observed macrophage phagocytosis deficit in chronic, non-healing wounds,13, 14, 15 the regulatory mechanisms underlying wound macrophage phagocytosis function are not fully deciphered. Differ from adhesion molecule β2 integrins, which mediate adhesion-dependent phagocytosis of apoptotic cells by wound macrophages,28, 31 PPARγ, as a nuclear transcription factor, transcriptionally regulates wound macrophage phagocytosis function. This finding may explain the poor wound healing in patients with chronic granulomatous disease, because macrophages from mice with chronic granulomatous disease have low levels of PPARγ expression,44 or poor wound healing in patients with other diseases present macrophage PPARγ low expression, and future studies are sure to test them.
Among all the tested macrophages phagocytosis-related molecules in our investigation, PPARγ consistently affected the expression of Cd36 and Mertk in PPARγ-KO macrophages. Although Cd36 is well known to be upregulated by PPARγ activation, the regulation of Mertk by PPARγ has been reported.45, 46 However, the expression of Mertk is induced by activating liver X receptor (LXR),47 and activating PPARγ will upregulate LXR,48 suggesting that Mertk expression may be induced by PPARγ via LXR.
TNF-α is a key factor impacting the healing process.35, 37 Excessive TNF-α expression in wound impaired the functions of keratinocytes, fibroblasts and endothelial cells,37, 49, 50 resulting in reduced reepithelialization, collagen deposition, angiogenesis and granulation tissue formation, and severely delayed wound healing.34, 36 Our results showed wound PPARγ-KO macrophages released excessive TNF-α, contributing to higher TNF-α expression in wounds, resulting in impaired wound healing in PPARγ-KO mice. Although in chronic, non-healing wounds, high expression of TNF-α by wound macrophages were observed, the underlying mechanisms are not fully known. However, the phagocytosis of apoptotic cells is associated with an anti-inflammatory response, such as suppressing TNF-α expression, in macrophages and the failure of apoptotic cell clearance is associated with chronic wound, suggesting the involvement of apoptotic cell engulfment in TNF-α expression regulation in skin wound. Here, we revealed that PPARγ regulates skin wound macrophages TNF-α expression by orchestrating their phagocytic function. It is well established that apoptotic cells are digested and cellular components, such as cholesterol and fatty acids, are released after engulfed by macrophages. These cellular components are ligands for PPARγ, which may induce PPARγ sumoylation to attenuate the removal of nuclear receptor corepressor from the κB site within the TNF-α promoter, resulting in blocking the transactivation of NF-κB and reducing TNF-α production in macrophages.51 Therefore, PPARγ can directly contribute to the TNF-α expression regulation induced by apoptotic cell engulfment. Correspondingly, the increased engulfment of apoptotic cells may enhance PPARγ activation to suppress TNF-α production. Our data here showed that PPARγ enhanced phagocytosis of apoptotic cells through increasing the expression of macrophage phagocytosis-related molecules, especially CD36 and Mertk, suggesting that PPARγ may also indirectly reduce TNF-α expression via upregulation of CD36 and Mertk.
Although our study here clearly showed that PPARγ was important for regulating wound macrophage phagocytosis function, the contribution of PPARγ to skin wound healing remains controversial. Previous studies showed that in fibroblasts, PPARγ agonists block TGF-β-induced αSMA, which is a myofibroblast differentiation marker and collagen expression,52, 53 and deletion of PPARγ in fibroblasts enhances dermal wound closure, indicating that PPARγ acts in fibroblasts retarding tissue repair. However, our results showed that PPARγ upregulated in wounded skin, which is consistent with previous study54 and in wound macrophages, and mice with macrophage PPARγ deficiency severely impaired wound healing, suggesting that PPARγ has a regulatory role in wound healing and acts in macrophages enhancing the healing process. Despite the controversial roles of PPARγ in macrophages and fibroblasts during wound healing, our data showed that PPARγ agonist accelerated the wound healing in WT but not PPARγ-KO mice, suggesting that PPARγ agonist accelerated wound healing because of activating macrophages PPARγ. So we concluded that activating PPARγ is helpful for improving skin wound healing.
In summary, our studies elucidated the pathophysiological mechanism of macrophage PPARγ in skin wound healing. After tissue injury, macrophage PPARγ is upregulated to promote timely disposal of apoptotic cells through increased the phagocytosis-related molecules expression, contributing to decreased local TNF-α expression to enhancing tissue repair (Figure 8d). Furthermore, we showed that activating PPARγ accelerated normal wound healing, providing the evidence of PPARγ may be a candidate for treatment of wound healing.
Materials and Methods
Mice
C57BL/6J male mice (8–12 weeks) were purchased from the Animal Center, Research Institute of Surgery and Daping hospital, TMMU, Chongqing, China. Mice carrying floxed alleles of PPARγ (PPARγf/f) and mice bearing the lysozyme-M Cre (LysMCre) recombinase transgene were purchased from the Jackson Laboratory (Mount Desert Island, ME, USA). PPARγf/f mice and LysMCre mice were crossed to generate offspring with macrophage-specific deficiency of the PPARγ gene. The PPARγf/fLysMCre− mice (PPARγ-WT) were used as WT control as previous reports and PPARγf/fLysMCre+ mice are referred to as PPARγ-KO mice.22, 55 All mice were on a C57BL/6 background and housed under a 12-h light–12-h dark cycle with free access to food and water. Wound-healing experiments were performed with male mice between 8 and 12 weeks of age. All animal procedures were in accordance with a protocol approved by the Local Administration District Official Committee. All efforts were made to minimize the number of animals and their suffering.
Analyses of macrophage-specific PPARγ deficient mice
The genotyping of determining PPARγ-WT and PPARγ-KO mice was performed by PCR of DNA obtained from tail biopsies. Primary peritoneal macrophages were recovered from mice injected with 3% thyoglicolate for 3 days. The mRNA levels and protein expression of PPARγ in both macrophage strains were performed by RT-PCR and WB. PPARγ expression in isolated wound macrophages and neutrophils, and splenic T cells, B cells and dendritic cells were analyzed by flow cytometry.
Wound-healing model
Full-thickness (including the panniculus carnosus) wounds were created in the dorsal skin under sterile conditions. Briefly, mice were anesthetized with peritoneal injection (i.p.) with pentobarbital sodium (50 mg/kg; Boster, Wuhan, China). After depilation with 8% Na2S and cleaning with povidone-iodine and 75% ethanol, the dorsal skin was picked up at the midline and punched with a sterile disposable biopsy punch (6 mm in diameter; Miltex, New York, NY, USA), generating one wound on the midline or two wounds on each side of the midline. One or four wounds per mice were induced. At indicated time points, each wound was digitally photographed, and wound areas were quantified using CorelDRAW 9 (Corel Software, Oakland, CA, USA). Changes in wound areas over time were expressed as the percentage of initial (day 0) wound area.
PVA sponges implantation and wound cell isolation
Mice were anesthetized with pentobarbital (50 mg/kg i.p.) and dorsal skin was depilated. Six PVA sponges (unlimited), measuring 1 × 1 × 0.6 cm, were inserted into individual s.c. pockets through a midline dorsal incision under sterile condition, and the skin was closed with clips.56 At specified times, mice were killed by CO2 asphyxiation, and the sponges were removed and a single-wound cell suspension was generated from sponges by repeated compression. The cell suspension was filtered through a 70 μm nylon cell strainer (Falcon, Shanghai, China) to remove all the sponge debris. When necessary, cells were subjected to red cell lysis buffer (Invitrogen, San Diego, CA, USA), followed by reconstitution in PBS. The s.c. sponge model is extensively used for wound-healing studies especially those investigating wound inflammation.57, 58, 59, 60 The primary difference between s.c. sponge model and excisional wound model is low-grade bacterial contamination of open wounds.61 Therefore, we routinely checked bacterial contamination in our excisional wounds using protocols described previously.62 As previous studies reported, although low-grade contamination was observed in superficial tissues, deep-tissue biopsies had not shown any bacterial contamination, suggesting that excisional deep-tissue wound cells are similar to that derived from PVA sponge.62
Total RNA isolation and quantitative RT-PCR
Total RNA was isolated using Tripure Isolation Reagent (Roche Group, Shanghai, China) and reverse transcribed into cDNA using Quantscript RT Kit (Tiangen Biotech, Beijing, China). The resulting cDNA was used to measure quantitatively the expression of genes using Quant Reverse Transcriptase according to the manufacturer’s protocol (Tiangen Biotech, Beijing, China). Real-time measurements of gene expression were performed with an iCycler thermocycler system and iQ5 optical system software (Bio-Rad Laboratory, Richmond, CA, USA). All primers were synthesized by Invitrogen Technologies (Carlsbad, CA, USA) and primer sequences used are presented as follows: β-actin (forward, 5′-TGGAATCCTGTGGCATCCATGAAA-3′; reverse, 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′), PPARγ (forward, 5′-TATCACTGGAGATCTCCGCCAACAGC-3′; reverse, 5′-GTCACGTTCTGACAGGACTGTGTGAC-3′), collagen type 1 (forward, 5′-ATCACTGCAAGAACAGCGTA-3′; reverse, 5′-TGTTTTCCAAAGTCCATGTG-3′), VEGF (forward, 5′-TGGATGTCTACCAGCGAAGC-3′; reverse, 5′-ACAAGGCTCACAGTGATTTT-3′), TNF-α (forward, 5′-AACTAGTGGTGCCAGCCGAT-3′; reverse, 5′-CTTCACAGAGCAATGACTCC-3′), Cd36 (forward, 5′-TCGGAACTGTGGGCTCATTG-3′; reverse, 5′-CCTCGGGGTCCTGAGTTATATTTTC-3′), Mertk (forward, 5′-GTGGCAGTGAAGACCATGAAGTTG-3′; reverse, 5′-GAACTCCGGGATAGGGAGTCAT-3′), Gas6 (forward, 5′-TCTTCTCACACTGTGCTGTTGCG-3′; reverse, 5′-GGTCAGGCAAGTTCTGAACACAT-3′), Mfge8 (forward, 5′-GGACATCTTCACCGAATACATCTGC-3′; reverse, 5′-TGATACCCGCATCTTCCGCAG-3′), C1qa (forward, 5′-AAAGGCAATCCAGGCAATATCA-3′; reverse, 5′-TGGTTCTGGTATGGACTCTCC-3′), C1qb (forward, 5′-AACGCGAACGAGAACTATGA-3′; reverse, 5′-ACGAGATTCACACACACAGGTTG-3′) and C1qc (forward, 5′-CAACGCCCTCGTCAGGTT-3′; reverse, 5′-ACAACCCAAGCACAGGGAAGT-3′). The RT-PCR data were analyzed by the method of 2−ΔΔCt and the data were normalized to β-actin.
Western blotting
Wound tissues and cultured cells were homogenized in ice-cold RIPA with 1 mg/ml of a protease inhibitor cocktail (Beyotime, Shanghai, China). The homogenates were centrifuged at 14 000 r.p.m. for 5 min at 4 °C and supernatants were collected. Subsequently, BCA assays were performed to determine the protein concentration of the supernatants. Standard protocol was used and following antibodies were applied: PPARγ (Abcam Inc., San Francisco, CA, USA), TNF-α (Santa Cruz Biotechnology, Santa Cruz, CA, USA), GAPDH (Goodhere Biotechnology, Hangzhou, China), the goat anti-rabbit IgG-HRP secondary antibody (Novus Biologicals, Littleton, CO, USA) and the goat anti-mouse IgG-HRP secondary antibody (Beyotime). The blots were detected by chemiluminescence with enhanced chemiluminescence reagent (BeyoECL Plus, Beyotime).
Histology
Wounded tissues were harvested, fixed in 4% paraformaldehyde, dehydrated, bisected, mounted in paraffin and sectioned for H&E staining, Masson trichrome staining and immunohistochemistry. Granulation tissues were stained using H&E. Collagen was stained with Masson trichrome. Immunohistochemistry was carried out with following antibodies: PPARγ (Abcam Inc.), F4/80 (Abcam Inc.), Ly-6G (eBioscience, San Diego, CA, USA), CD31 (Boster), the goat anti-rat IgG-biotin secondary antibody (Invitrogen) and the goat anti-rabbit IgG-biotin secondary antibody (Invitrogen). The protocol used for immunohistochemistry was performed as reported previously.63 Briefly, paraffin-embedded tissue blocks were cut into 4 μm sections. After the sections were dewaxed and rehydrated, antigen retrieval was performed by microwaving in citrate buffer (pH 6.0). The sections were cooled to room temperature (RT), and endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxidase (H2O2) for 10 min. The sections were incubated with 3% albumin boving V to block nonspecific binding at RT. Thereafter, the sections were incubated with primary antibodies overnight at 4 °C. After washing, the sections were incubated with corresponding secondary antibodies for 30 min at RT. Subsequently, the Vecta-stain ABC kit (Vector Laboratories, San Diego, CA, USA) was used for the avidin–biotin complex method according the manufacturer’s instruction. Peroxidase activity was visualized with DAB Elite kit (K3465, DAKO, Copenhagen, Denmark). The sections were lightly counterstained with hematoxylin and dehydrated through an ethanol series to xylene and mounted.
Tunel staining was performed using a commercially available kit (Derma TACS, Trevigen Inc., Gaithersburg, MD, USA).
All the sections were viewed using a light and fluorescence microscope. For image quantification, 3–5 high-powered images were quantified for each data point per animal.
Local TNF-α restoration and PPARγ agonist treatment
In TNF-α restoration experiments, wound margins were s.c. injected with 10 μg per wound purified aTNF-α (Sungene, Tianjin, China) on days 3, 4, 5, 6 and 7 after wounding. Mice treated with isotype control antibody (Sungene) served as mock controls. For PPARγ agonist RSG (Sigma Aldrich, St. Louis, MO, USA) treatment, mice were given RSG (10 mg/kg) or vehicle (carboxymethyl cellulose) (Sangon Biotech, Shanghai, China) via oral gavage for 2 days before wounding and every day thereafter until day 12 after wounding.
Gene expression and cytokine secretion assays
Primary peritoneal macrophages were collected from abdomen of mice, which were injected with 3% thyoglicolate for 3 days. Cells were incubated in RPMI 1640 medium supplemented with 10% FCS. To generate ATs, thymi from 3- to 4- week-old C57BL/6 mice were harvested and ground, filtered, pelleted and resuspended in RPMI 1640 medium supplemented with 10% FCS. Apoptosis was induced by treatment with 1 μM of dexamethasone (Sigma Aldrich) for 6 h. For AT assays, ATs were added to macrophages (5 : 1) for 60 min or 24 h. When needed, ATs were removed by gently washing, and macrophages were incubated with 100 ng/ml LPS for additional 5 h for gene expression analysis and 18 h for cytokine secretion studies. For ligand assays, macrophages were incubated with 100 nM RSG for 24 h. Gene expression was performed by RT-PCR and cytokine secretion was quantified in culture supernatants using commercially available ELISA kits (BD Biosciences, Franklin Lakes, NJ, USA).
In vitro phagocytosis assays
CFSE-labeled ATs were added to peritoneal macrophages in a 5 : 1 ratio and co-cultured in RPMI supplemented with 10% FBS at 37 °C for 60 min. After incubation with ATs, macrophages were gently washed several times with cold PBS and Cell Dissociation Buffer, Enzyme Free PBS-based (Invitrogen). A single-cell suspension of macrophages was stained with PE-conjugated anti-mouse F4/80 antibody (Sungene Biotech, Tianjin, China) and cells were analyzed by flow cytometry.
Flow cytometric analyses of wound cells isolated from PVA sponges
Wound cells were isolated from PVA sponges at indicated time points. Antibodies used for cells staining included: Ly-6G, F4/80, CD11b, annexin V, anti-rabbit IgG (FITC-conjugated secondary antibody) all from Sungene Biotech; PPARγ from Abcam Inc.; TNF-α and normal rabbit IgG from Santa Cruz Biotechnology. Cells were analyzed with a FACScan (BD Biosciences). Flow cytometry analysis was done with the FlowJo software (FlowJo, Hangzhou, China).
Flow cytometric analyses of splenic T cells, B cells and dendritic cells
The spleen was cut from abdomen and ground, and erythrocytes were lysed with red cell lysis buffer. The cells were then resuspended in PBS. Antibodies used for cells staining included: CD3, CD4, CD8, CD19, CD11b, CD11c, anti-rabbit IgG (FITC-conjugated secondary antibody) all from Sungene Biotech; PPARγ from Abcam Inc.; normal rabbit IgG from Santa Cruz Biotechnology. Cells were analyzed with a FACScan. Flow cytometry analysis was done with the FlowJo software.
Statistical analysis
Data were statistically analyzed using GraphPadPrism 5.0 (GraphPad Software, Beijing, China) and expressed as the mean±S.D. for the indicated number of independently performed duplicated experiments. Statistical significance was determined by two-tailed Student’s t-test. For all statistical analyses, significance levels were set at P-values <0.05.
Abbreviations
- ATs:
-
apoptotic thymocytes
- KO:
-
knock out
- LPS:
-
lipopolysaccharide
- PPARγ:
-
peroxisome proliferator-activated receptor gamma
- RSG:
-
rosiglitazone
- TNF-α:
-
tumor necrosis factor alpha
- WT:
-
wild type
References
Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH . Macrophages in skin injury and repair. Immunobiology 2011; 216: 753–762.
Li J, Chen J, Kirsner R . Pathophysiology of acute wound healing. Clin Dermatol 2007; 25: 9–18.
Singer AJ, Clark RA . Cutaneous wound healing. N Engl J Med 1999; 341: 738–746.
Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 2009; 17: 763–771.
Natarajan S, Williamson D, Stiltz AJ, Harding K . Advances in wound care and healing technology. Am J Clin Dermatol 2000; 1: 269–275.
Martin JM, Zenilman JM, Lazarus GS . Molecular microbiology: new dimensions for cutaneous biology and wound healing. J Invest Dermatol 2010; 130: 38–48.
Mirza R, DiPietro LA, Koh TJ . Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol 2009; 175: 2454–2462.
Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol 2010; 184: 3964–3977.
Koh TJ, DiPietro LA . Inflammation and wound healing: the role of the macrophage. Exp Rev Mol Med 2011; 13: e23.
Stout RD . Editorial: macrophage functional phenotypes: no alternatives in dermal wound healing? J Leukoc Biol 2010; 87: 19–21.
Wu YS, Chen SN . Apoptotic cell: linkage of inflammation and wound healing. Front Pharmacol 2014; 5: 1.
Rodero MP, Khosrotehrani K . Skin wound healing modulation by macrophages. Int J Clin Exp Pathol 2010; 3: 643–653.
Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PloS One 2010; 5: e9539.
Tang Y, Zhang MJ, Hellmann J, Kosuri M, Bhatnagar A, Spite M . Proresolution therapy for the treatment of delayed healing of diabetic wounds. Diabetes 2013; 62: 618–627.
Swift ME, Burns AL, Gray KL, DiPietro LA . Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol 2001; 117: 1027–1035.
Michalik L, Desvergne B, Tan NS, Basu-Modak S, Escher P, Rieusset J et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J Cell Biol 2001; 154: 799–814.
Tan NS, Michalik L, Desvergne B, Wahli W . Peroxisome proliferator-activated receptor (PPAR)-beta as a target for wound healing drugs: what is possible? Am J Clin Dermatol 2003; 4: 523–530.
Tan NS, Michalik L, Di-Poi N, Desvergne B, Wahli W . Critical roles of the nuclear receptor PPARbeta (peroxisome-proliferator-activated receptor beta) in skin wound healing. Biochem Soc Transac 2004; 32: 97–102.
Li J, Li P, Zhang Y, Li GB, He FT, Zhou YG et al. Upregulation of ski in fibroblast is implicated in the peroxisome proliferator—activated receptor delta-mediated wound healing. Cell Physiol Biochem 2012; 30: 1059–1071.
Rigamonti E, Chinetti-Gbaguidi G, Staels B . Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol 2008; 28: 1050–1059.
Majai G, Sarang Z, Csomos K, Zahuczky G, Fesus L . PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol 2007; 37: 1343–1354.
Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol 2011; 186: 621–631.
Asada K, Sasaki S, Suda T, Chida K, Nakamura H . Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am J Respir Crit Care Med 2004; 169: 195–200.
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007; 447: 1116–1120.
Low QE, Drugea IA, Duffner LA, Quinn DG, Cook DN, Rollins BJ et al. Wound healing in MIP-1alpha(-/-) and MCP-1(-/-) mice. Am J Pathol 2001; 159: 457–463.
Martin P, D'Souza D, Martin J, Grose R, Cooper L, Maki R et al. Wound healing in the PU.1 null mouse—tissue repair is not dependent on inflammatory cells. Curr Biol 2003; 13: 1122–1128.
Martin P, Leibovich SJ . Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 2005; 15: 599–607.
Peters T, Sindrilaru A, Hinz B, Hinrichs R, Menke A, Al-Azzeh EA et al. Wound-healing defect of CD18(-/-) mice due to a decrease in TGF-beta1 and myofibroblast differentiation. EMBO J 2005; 24: 3400–3410.
Maruyama K, Asai J, Ii M, Thorne T, Losordo DW, D'Amore PA . Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am J Pathol 2007; 170: 1178–1191.
Ishida Y, Gao JL, Murphy PM . Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J Immunol 2008; 180: 569–579.
Sindrilaru A, Peters T, Schymeinsky J, Oreshkova T, Wang H, Gompf A et al. Wound healing defect of Vav3-/- mice due to impaired {beta}2-integrin-dependent macrophage phagocytosis of apoptotic neutrophils. Blood 2009; 113: 5266–5276.
Goldberg MT, Han YP, Yan C, Shaw MC, Garner WL . TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: an implication for abnormal wound healing. J Invest Dermat 2007; 127: 2645–2655.
Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 2010; 53: 378–388.
Lai JJ, Lai KP, Chuang KH, Chang P, Yu IC, Lin WJ et al. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J Clin Invest 2009; 119: 3739–3751.
Ashcroft GS, Jeong MJ, Ashworth JJ, Hardman M, Jin W, Moutsopoulos N et al. Tumor necrosis factor-alpha (TNF-alpha) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen 2012; 20: 38–49.
Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 2011; 121: 985–997.
Xu F, Zhang C, Graves DT . Abnormal cell responses and role of TNF-alpha in impaired diabetic wound healing. BioMed Res Int 2013; 2013: 754802.
Werner S, Grose R . Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003; 83: 835–870.
Weigert A, Jennewein C, Brune B . The liaison between apoptotic cells and macrophages—the end programs the beginning. Biol Chem 2009; 390: 379–390.
Hart SP, Smith JR, Dransfield I . Phagocytosis of opsonized apoptotic cells: roles for ‘old-fashioned’ receptors for antibody and complement. Clin Exp Immunol 2004; 135: 181–185.
Kliewer SA, Xu HE, Lambert MH, Willson TM . Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res 2001; 56: 239–263.
Erwig LP, Henson PM . Clearance of apoptotic cells by phagocytes. Cell Death Differ 2008; 15: 243–250.
Galli SJ, Borregaard N, Wynn TA . Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 2011; 12: 1035–1044.
Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 2009; 113: 2047–2055.
Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM Jr ., Hajjar DP et al. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res 2000; 41: 688–696.
McCarty MF, Barroso-Aranda J, Contreras F . PPAR gamma agonists can be expected to potentiate the efficacy of metronomic chemotherapy through CD36 up-regulation. Med Hypotheses 2008; 70: 419–423.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009; 31: 245–258.
Piraino G, Cook JA, O'Connor M, Hake PW, Burroughs TJ, Teti D et al. Synergistic effect of peroxisome proliferator activated receptor-gamma and liver X receptor-alpha in the regulation of inflammation in macrophages. Shock 2006; 26: 146–153.
Salomon GD, Kasid A, Cromack DT, Director E, Talbot TL, Sank A et al. The local effects of cachectin/tumor necrosis factor on wound healing. Ann Surg 1991; 214: 175–180.
Rapala K, Laato M, Niinikoski J, Kujari H, Soder O, Mauviel A et al. Tumor necrosis factor alpha inhibits wound healing in the rat. Eur Surg Res 1991; 23: 261–268.
Jennewein C, Kuhn AM, Schmidt MV, Meilladec-Jullig V, von Knethen A, Gonzalez FJ et al. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J Immunol 2008; 181: 5646–5652.
Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol 2005; 288: L1146–L1153.
Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J . Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am J Pathol 2009; 174: 519–533.
Kapoor M, Kojima F, Yang L, Crofford LJ . Sequential induction of pro- and anti-inflammatory prostaglandins and peroxisome proliferators-activated receptor-gamma during normal wound healing: a time course study. Prostag Leukotr Ess Fatty Acids 2007; 76: 103–112.
Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest 2007; 117: 1658–1669.
Meszaros AJ, Reichner JS, Albina JE . Macrophage phagocytosis of wound neutrophils. J Leukoc Biol 1999; 65: 35–42.
Albina JE, Mills CD, Barbul A, Thirkill CE, Henry Jr WL, Mastrofrancesco B et al. Arginine metabolism in wounds. Am J Physiol 1988; 254: E459–E467.
Efron DT, Barbul A . Subcutaneous sponge models. Methods Mol Med 2003; 78: 83–93.
Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE . The phenotype of murine wound macrophages. J Leukoc Biol 2010; 87: 59–67.
Daley JM, Reichner JS, Mahoney EJ, Manfield L, Henry Jr WL, Mastrofrancesco B et al. Modulation of macrophage phenotype by soluble product(s) released from neutrophils. J Immunol 2005; 174: 2265–2272.
Schaffer MR, Tantry U, Barbul A . Wound fluid inhibits wound fibroblast nitric oxide synthesis. J Surg Res 2004; 122: 43–48.
Roy S, Khanna S, Nallu K, Hunt TK, Sen CK . Dermal wound healing is subject to redox control. Mol Ther 2006; 13: 211–220.
Li H, Wang C, Guo G, Gao C, Wu Y, Chen Y . The characteristic expression of B7-associated proteins in Langerhans cell sarcoma. Acta Histochem 2012; 114: 733–743.
Acknowledgements
This investigation was supported by the National Natural Science Foundation of China (31170851 and 81471570) and the SRF for ROCS, SEM.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Aberdam
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International Licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, H., Shi, R., Luo, B. et al. Macrophage peroxisome proliferator-activated receptor γ deficiency delays skin wound healing through impairing apoptotic cell clearance in mice. Cell Death Dis 6, e1597 (2015). https://doi.org/10.1038/cddis.2014.544
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.544
This article is cited by
-
Preadipocytes in human granulation tissue: role in wound healing and response to macrophage polarization
Inflammation and Regeneration (2023)
-
Macrophage mediation in normal and diabetic wound healing responses
Inflammation Research (2020)
-
Development of Novel Mouse Model of Ulcers Induced by Implantation of Magnets
Scientific Reports (2017)
-
Phagocyte respiratory burst activates macrophage erythropoietin signalling to promote acute inflammation resolution
Nature Communications (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}