
Abstract
Phenothiazines are a family of heterocyclic compounds whose clinical utility includes treatment of psychiatric disorders as well as chemotherapy-induced emesis. Various studies have demonstrated that these compounds possess cytotoxic activities in tumor cell lines of different origin. However, there is considerable confusion regarding the molecular basis of phenothiazine-induced cell death. Lung cancer (LC) remains one of the most prevalent and deadly malignancies worldwide despite considerable efforts in the development of treatment strategies, especially new targeted therapies. In this work, we evaluated the potential utility of phenothiazines in human LC. We show that phenothiazines as single treatment decreased cell viability and induced cell death preferentially in small cell lung carcinoma (SCLC) over non small cell lung carcinoma (NSCLC) cell lines. Sensitivity to phenothiazines was not correlated with induction of apoptosis but due to phenothiazine-induced lysosomal dysfunction. Interestingly, the higher susceptibility of SCLC cells to phenothiazine-induced cell death correlated with an intrinsically lower buffer capacity in response to disruption of lysosomal homeostasis. Importantly, this effect in SCLC occurred despite mutation in p53 and was not influenced by intrinsic sensitivity/resistance toward conventional chemotherapeutic agents. Our data thus uncovered a novel context-dependent activity of phenothiazines in SCLC and suggest that phenothiazines could be considered as a treatment regimen of this disease, however, extended cell line analyses as well as in vivo studies are needed to make such conclusion.
Similar content being viewed by others


Main
Phenothiazines are a family of heterocyclic compounds whose clinical utility includes treatment of psychiatric disorders, for example, schizophrenia.1 In this setting, the therapeutically relevant targets of phenothiazines are believed to be neurotransmitter receptors, in particular the dopamine D2 receptor (D2R).2 However, an array of putative cellular targets have additionally been described for phenothiazines, ranging from calmodulin (CaM) to lipid membranes, suggesting that the effect of these compounds likely extend beyond their impact on neuroendocrine signaling.3 Notably, various studies have demonstrated that phenothiazines possess cytotoxic activities, especially in established tumor cell lines.4 However, there is considerable confusion regarding the molecular basis of phenothiazine-induced cell death and numerous hypotheses, including CaM antagonism,5 membrane fluidization6 and disruption of mitosis,7 have been proposed.
Lung cancer (LC) remains one of the most deadly malignancies worldwide8 despite considerable effort in the development of treatment strategies, especially new targeted therapies. In parallel, there has been a recent revival of interest in the use of ‘old’ FDA-approved drugs for new indications. In this work, we evaluated the potential utility of phenothiazines in LC. We show that phenothiazines induced cell death preferentially in small cell lung carcinoma (SCLC) over non small cell lung carcinoma (NSCLC) cell lines. In this context, cytotoxicity was due to phenothiazine-induced lysosomal dysfunction and the higher susceptibility of SCLC cells correlated with an intrinsically lower buffer capacity in response to disruption of lysosomal homeostasis. Importantly, this effect in SCLC occurred despite mutation in p53 and was not influenced by intrinsic sensitivity/resistance toward the conventional chemotherapeutic agents, for example, cisplatin, etoposide or gemcitabine. Our data thus uncovered a novel context-dependent antitumor action of phenothiazines in SCLC.
Results
Single-drug treatment with phenothiazines induced cytotoxicity preferentially in human SCLCs
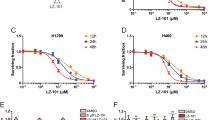
Phenothiazines have been reported to induce apoptosis in tumor cells of different origin.3, 4, 9 Here, we evaluated the potential therapeutic utility of phenothiazines in a panel of human SCLC and NSCLC cell lines. At a concentration of 10 μM, which is in the range of concentration achievable in the plasma of patients without eliciting significant adverse side effects,10 phenothiazines were overtly cytotoxic, analyzed as reduction in cell viability, in most of the tested SCLC cell lines while NSCLC cell lines were generally less responsive (Figures 1a–c, Table 1). The hyper-responsiveness of SCLC to phenothiazines was not recapitulated when the chemotherapeutic drugs cisplatin, etoposide and gemcitabine were used,11 as comparable decrease in cell viability was observed in SCLC and NSCLC cells (Supplementary Figure S1). Moreover, our results clearly show that the sensitivity to phenothiazine-induced decrease in cell viability in SCLC is not substantially influenced by growth pattern, site of isolation, prior exposure to conventional chemotherapies and occurs despite mutation in the tumor suppressor p53 (Supplementary Table S1). To further verify the preferential activity of phenothiazines in SCLC, we compared the sensitivity of four phenothiazine compounds (trifluoperazine dihydrochloride (TFP), fluphenazine dihydrochloride (FPZ), triflupromazine hydrochloride (TFPZ) and promazine hydrochloride (PZ)) over a range of concentrations in the SCLC cell line H82 and in the NCSLC cell line U-1810 (Figure 1b, upper panel).

Phenothiazines are preferentially cytotoxic in human SCLC cell lines. (a) Top panel: Cell viability of SCLC and NSCLC cell lines after treatment with six different phenothiazines (10 μM fixed dose, 72 h); cell viability was measured by MTT and untreated cells were set to 100%. Bottom panel: Mean cell viability of WI-38, NSCLC or SCLC cell lines treated with TFP, FPZ, PZ and TFPZ (10 μM, 72). *denotes difference in sensitivity between NSCLC and SCLC cell line panels, unpaired two-tailed t-test P=0.0003. (b) Upper panel, H82 (SCLC) and U-1810 (NSCLC) were exposed to different doses of phenothiazines for 72 h. Lower panel, H69, U-1285, U-1906, U-2020 (all SCLC) and A549, H125, H1299 (all NSCLC) cells were treated with the indicated concentrations of TFP for 72 h. Cell viability was measured by MTT and untreated cells set to 1. (c) WI-38 (normal lung fibroblasts) was exposed to different doses of phenothiazines for 72 h; cell viability was measured by MTT. Data depict mean±S.D. compiled from three independent experiments
We also assessed the dose-dependent cytotoxicity of TFP, analyzed as decrease in cell viability, in several additional SCLC (H69, U-1285, U-1906 and U-2020) and NSCLC (A549, H125 and H1299) cell lines (Figure 1b, lower panel). These analyses confirmed that SCLC cells were more sensitive to phenothiazines than NSCLC cells (Figure 1b). Importantly, the cell viability of primary fetal lung WI-38 fibroblast was less affected by phenothiazines at concentrations that caused significant cytotoxicity in SCLC cells, illustrating a potential therapeutic window for phenothiazines in SCLC (Figures 1a and c). The increased sensitivity of SCLC cells to phenothiazines was also evidenced by increased cell death and growth arrest after treatment with these agents (Figure 2). Thus, 10 μM TFP induced a prominent impairment in cell division capacity in SCLC cells, while in NSCLC cells 20 μM TFP was needed to significantly affect proliferation (Figures 2a and b). Analysis of cell cycle kinetics of SCLC cells (H69 and H82) revealed that 10 μM TFP caused a time-dependent cell cycle arrest in late S and G2/M phases, whereas such changes in cell cycle were not observed for NSCLC (U-1810; Figure 2c). Taken together, our data demonstrate that phenothiazines preferentially decrease the viability and growth of human SCLC.

TFP impairs cell proliferation. (a) Morphology of H82 and U-1810 cells were examined under a light microscope after treatment with TFP at the indicated concentrations for 72 h. (b) H82 and U-1810 cells were treated with TFP at the indicated concentrations for 48 h; the number of cell division was estimated by CFSE retention. Data depict mean±S.D. compiled from three independent experiments. Asterisk indicates a statistically significant difference (P<0.05, two-tailed t-test) compared with mock treatment. (c) H69 and H82 cells were treated with TFP at the indicated concentration for 6, 24 or 48 h. For comparison U-1810 cells were treated with 10 μM TFP for 48 h. DNA content analysis was performed by PI staining and cell cycle distribution was assigned by ModFit. Data shown are representative of three independent experiments
Phenothiazines elicit caspase-mediated and caspase-independent cell death
To gain insight into the mechanistic basis of phenothiazine-associated cytotoxicity, we analyzed in detail the mode by which these compounds induce cell death in human SCLC cells. While some studies have implicated apoptosis as a major cell death mode in phenothiazine-treated cells,3, 4, 9 we found that the percentage of SCLC exhibiting nuclear morphologic changes typical of apoptosis, such as chromatin condensation and fragmentation, remained low (10%) even at the highest TFP concentrations, where >98% of all cells lost the ability to exclude propidium iodide (PI; data not shown). Instead, TFP-treated SCLC cells exhibited profoundly shrunken nuclei without concomitant chromatin condensation. TFP elicited poly (ADP-ribose) polymerase (PARP) cleavage in some, but not all, LC cell lines (Figures 3a and b). Importantly, although the SCLC cells are sensitive to 10 μM TFP treatment and respond with an approximately 60% decrease in cell viability, this concentration of TFP only resulted in no or minor cleavage of PARP. Consistent with a non-essential requirement of caspase-mediated apoptotic response, the pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp fluoromethylketone (z-VAD-fmk) did not significantly protect either SCLC (H82) or NSCLC (U-1810) cells from TFP-induced cell death (Figure 3c), emphasizing the involvement of non-caspase-mediated cell death in response to phenothiazines in SCLC.

TFP induces cell death distinct from classical apoptosis. (a) SCLC (H82, H69) and NSCLC (U-1810, A549) cells were treated with TFP at the indicated concentrations for 6, 24 or 48 h; WCL was used for immunoblotting with an antibody recognizing full-length and cleaved PARP (113 and 89 kDa respectively). GAPDH antibody was used to confirm equal loading. (b) H592, U-1285, H125, H157 cells were treated with TFP at the indicated concentrations for 72 h; immunoblotting was performed as in (a). (c) H82 and U-1810 cells were pre-treated (1 h) with z-VAD-fmk (20 μM) and thereafter exposed to TFP at the indicated concentrations for 72 h; cell viability was measured by MTT. For (a and b), data shown are representative of three independent experiments. For (c), data depict mean±S.D. compiled from three independent experiments. NS, nonsignificant
Phenothiazines induce lysosomal dysfunction in LC cells
Phenothiazines are known to be lysosomotropic and accumulate within intracellular acidic compartments.12, 13 We therefore examined whether phenothiazines may affect lysosomal functions in LC cells. We found that treatment with TFP and related phenothiazines (cis-flupenthixol dihydrochloride (cis-FPX), PZ, chlorpromazine hydrochloride (CPZ), TFPZ and FPZ) caused a rapid increase in appearance of light chain 3 (LC3)-II, a marker of autophagy, in a dose- and time-dependent manner in both SCLC and NSCLC cells (Figures 4a–c). By contrast, the level of p62/SQSTM1, a scaffold protein that target ubiquitinated polypeptide cargo for autophagic degradation, did not show any consistent change in response to TFP in the examined SCLC and NSCLC cells (Figure 4a). As accumulation of LC3-II can result from either increased autophagic flux or impaired autophagic degradation, we used chemical probes to manipulate these two processes separately and then investigated the effect of phenothiazines on LC3 conversion. Notably, blocking autophagic degradation with the lysosomal protease inhibitor E-64d in cells treated with survivable concentrations of TFP (5 μM in H82 and 10 μM in U-1810) caused further accumulation of LC3-II, indicative of enhanced autophagic flux (Figure 4d). Similar results were obtained at cytotoxic concentrations of TFP or when autophagosome maturation was suppressed by bafilomycin A1 (BafA1; data not shown). Under conditions where de novo protein synthesis was shut down by cycloheximide (CHX), LC3-II induced by TFP pre-treatment (0–24 h) was rapidly cleared upon removal of TFP (Figure 4e, compare lanes 2 and 4). This process became significantly slower if TFP was present during the recovery period (24–48 h), especially in H82 cells (Figure 4e, compare lanes 5 and 6), suggesting that phenothiazines may additionally antagonize autophagic degradation. These data show that TFP both induces LC3-II and prevents its clearance, especially in the SCLC cells, which is consistent with the notion that phenothiazines perturb lysosome homeostasis more severely and persistently in SCLC than in NSCLC. Overall, these data demonstrate that phenothiazines can modulate lysosomal functions in human LC cells.

Phenothiazines disrupt autophagy. (a) H82, H69, U-1810 and A549 cells were treated with TFP at the indicated concentrations for 6, 24 or 48 h; WCL was used for immunoblotting with antibodies specific for p62, LC3B (which detect both LC3-I and LC3-II) and GAPDH (loading control). (b) H592, U-1285, H125, H157 cells were treated with TFP at the indicated concentrations for 72 h; WCL was used for immunoblotting with antibodies as in (a). (c) H82 and U-1810 cells were treated with 10 μM of the indicated phenothiazines for 24 h; WCL was used for immunoblotting with antibodies specific for LC3B and GAPDH (loading control). (d) H82 and U-1810 cells were treated with TFP at the indicated concentrations in the absence or presence of E-64d (30 μM) for 24 h; WCL was used for immunoblotting as in (a). (e) H82 and U-1810 cells were treated with TFP for 24 h followed by 24 h with either TFP, cycloheximide (CHX, 100 μg/ml) or TFP+CHX. WCL was used for immunoblotting as in (a). Data shown are representative of three independent experiments
Basal lysosomal mass and pH buffer capacity can predict sensitivity to phenothiazines
The response to chemotherapy (CT) in human tumors is highly heterogenous and therefore there is an urgent need for the identification of biomarkers for prediction of treatment efficacy. As our data suggest that the lysosome is a potentially critical site of action for phenothiazines when used as single treatment of LC cells, we therefore sought to determine whether lysosome-associated parameters could be used to predict sensitivity of human LC cells to phenothiazines. Indeed, we found a statistically significant inverse correlation between the mean cytotoxicity index, calculated from the average cytotoxicity of six phenothiazines at 10 μM (Figure 1a, Table 1), and cellular retention of LysoTracker at baseline (Figure 5a). On the other hand, neither basal lysosomal β-galactosidase (β-gal) activity nor the baseline expression levels of the lysosomal markers LC3-II and lysosomal-associated membrane protein 1 (LAMP-1) correlated well with cellular phenothiazine sensitivity (Supplementary Figures S2A–C). As differential LysoTracker retention may reflect differences in basal lysosomal mass and/or lysosomal pH, we therefore next examined phenothiazine-induced changes in LysoTracker retention (ΔLysoTracker) as an indicator of perturbation in lysosomal pH. Exposure to TFP for 24 h was associated with varying degrees of loss in LysoTracker retention in almost all of the LC cell lines tested (Figure 5b). Furthermore, there was a highly significant positive correlation between the extent of TFP-induced loss in LysoTracker retention and the magnitude of cytotoxicity (Figure 5b, left). Importantly, the loss of LysoTracker retention became irreversible in SCLC at lower TFP concentrations than in NSCLC cells (10 μM in H82 and 20 μM in U-1810) (Figure 5c), and the afflicted cells underwent clear mitochondrial depolarization (Figure 5d). By contrast, cells exposed to non-cytotoxic concentrations of TFP recovered LysoTracker Green staining to various degrees over time (Figure 5c). These data showed that prolonged disruption of lysosomal functions was detrimental to LC cell viability and that SCLC were more sensitive to phenothiazines as result of lower intrinsic lysosomal buffer capacity. In line with these results, TFP-induced cell death was significantly antagonized by the protease inhibitor E-64d (Figure 5e). Moreover, preventing the accumulation of TFP within lysosomes by BafA1 pre-treatment also largely abolished its negative impact on cell viability (Figure 5f). Thus, our data suggest that the more prominent cytotoxic effect of phenothiazines in SCLC is related to a higher degree of lysosomal pH neutralization (ΔLysoTracker) that is more persistent in SCLC than in NSCLC. Moreover, although phenothiazines initially induced LC3-II in both NSCLC and SCLC, only SCLC failed to adapt to phenothiazine-induced lysosomal dysfunction and this led to increased cell death preferentially in SCLC. Together, these data reinforce the notion that persistent lysosomal perturbation induced upon single-agent phenothiazine treatment is a major mediator of cytotoxicity in SCLC cells.

Baseline and TFP-induced loss of LysoTracker retention correlate with phenothiazine sensitivity. (a) Eight SCLC (1: H69, 2: H82, 3: H592, 4: U-1285, 5: U-1568, 6: U-1690, 7: U-1906, 8: U-2020) and eight NSCLC (9: A549, 10: H125, 11: H1299, 12: H157, 13: H23, 14: H661, 15: U-1752, 16: U-1810) cell lines were collected without treatment and analyzed for baseline LysoTracker retention; correlation between baseline LysoTracker retention (log) and phenothiazine toxicity (see Figure 1) is shown on the right. (b) Cells were treated with 10 μM TFP for 24 h and thereafter analyzed for TFP-induced changes in LysoTracker retention (ΔLysoTracker); correlation between ΔLysoTracker and phenothiazine toxicity is shown on the right. (c) H82 and U-1810 cells were treated with TFP at the indicated concentrations for 6, 24 or 48 h; lysosomal pH was estimated by LysoTracker retention. (d) H82 and U-1810 cells were treated with TFP at the indicated concentrations for 6, 24 or 48 h; mitochondrial membrane potential was estimated by TMRE retention. (e) H82 and U-1810 cells were pre-treated (1 h) with E-64d (30 μM) and thereafter exposed to TFP at the indicated concentrations for 72 h; cell viability was measured by MTT and untreated cells were set to 1. (f) Same as in (e), except cells were pre-treated with BafA1 (1 nM). Data depict mean±S.D. compiled from three independent experiments. Asterisk indicates a statistically significant difference (P<0.05, two-tailed t-test)
SCLC cells show increased sensitivity toward lysosome-disrupting agents
On the basis of the findings presented above, we reasoned that the increased susceptibility of SCLC cells to phenothiazines may be due to lower basal lysosomal mass and/or lysosomal pH buffer capacity, leading to intrinsically lower tolerance toward compounds that disrupt lysosomal functions. Consistent with this idea, we found that SCLC cells were typically more sensitive than NSCLC counterparts toward a number of lysosomotropic agents (Supplementary Figure S3), including the clinically approved drugs tamoxifen (TMX) and chloroquine (CQ), in addition to the phenothiazine compounds TFP and CPZ. Notably, TFP, CPZ, TMX and CQ all dissipated LysoTracker staining to a larger extent in SCLC (H69, H82, H592 and U-1285) than in NSCLC (A549 and U-1810) cell lines (Figures 6a and b). Moreover, the increased sensitivity of SCLC cells toward lysosomotropic agents was not recapitulated with the CaM antagonist W7, suggesting that phenothiazine-induced lysosomal perturbation does not involve modulation of Ca2+/CaM signaling, a well-known process inhibited by phenothiazines (Figure 6c, Supplementary Figure S3A). Overall, our findings highlight not only the potential for treating SCLC with FDA-approved drugs that target lysosomal acidification in general, but also the use of phenothiazines against this tumor type in particular.

SCLC cells are more susceptible to lysosomal perturbation but not calmodulin antagonism. (a and b) H69, H82, H592, U-1285, A549 and U-1810 cells were treated with 10 μM TFP or 10 μM CPZ (a), 10 μM TMX or 20 μg/ml CQ (b) for 24 or 48 h and thereafter analyzed for drug-induced changes in LysoTracker retention. (c) Similar to (a and b) except cells were treated with 10 μM W7. Data depict mean±S.D. compiled from at least three independent experiments
Discussion
Phenothiazines are most well-known for their dopamine antagonistic activity in the central nervous system (CNS), which has been utilized clinically for the management of psychiatric disorders such as schizophrenia.1, 2 In addition, these compounds affect the fate of non-CNS cells in a variety of ways, and depending on the experimental context, may result in cellular differentiation, cell death or protection from toxic injuries.3, 4, 9 One well-recognized property of phenothiazines is their ability to induce apoptosis in certain cell types.3, 4, 9 The mechanism(s) behind this is not clearly understood, although inhibition of CaM-regulated processes and increases in membrane fluidity/permeability are thought to be at least partially responsible. However, the cell-intrinsic determinants that govern which of these effects that will be elicited in a particular model system are not known, and the underlining molecular pathways remain poorly defined. Elucidating the contextual utilities of phenothiazines in in vitro system, response in tumor xenografts and animal models will enable them to be harnessed appropriately for treating different human ailments, including tumors, as illustrated here with SCLC.
Although targeted agents have been introduced for the clinical management of LC cases, the majority of SCLC and NSCLC patients with advanced disseminated diseases are still treated with conventional CT agents such as cisplatin. For SCLC, the initial response is often good but most cases relapse and present high degrees of chemoresistance, while for NSCLC a less CT-sensitive phenotype is usually found already at start of treatment.14 Consequently, there is an urgent need for the development of new treatment regimen to combat both subtypes of LC. In this study, we evaluated a novel strategy involving the use of phenothiazines as single treatment agents in LC. Our data demonstrate that phenothiazines are generally more cytotoxic in SCLC than in NSCLC cell lines despite comparable responses to cisplatin, etoposide and gemcitabine, which are standard chemotherapeutic agents for the treatment of LC. Importantly, we show that normal lung fibroblasts are less affected by phenothiazines at concentrations, which were toxic for SCLC indicating a favorable therapeutic window that would allow its use in SCLC without incurring significant adverse effects on healthy tissues. Although it needs to be confirmed by further in vivo toxicity analysis; several earlier reports have shown that phenothiazines are in general well-tolerated by cancer patients.10, 15 To uncover responsible mechanisms for the preferential susceptibility of SCLC to phenothiazines, we dissected the molecular details of phenothiazine-induced cell death using multiple SCLC and NSCLC cell lines and with TFP as a model compound. We found that TFP treatment led to a rapid neutralization of lysosomal pH, as judged by decreased retention of the lysosomotropic dye LysoTracker Green, accompanied by accumulation of LC3-II in SCLC cells. Our data thus corroborated a previous study that identified TFP as an inducer of autophagy at low doses in H4 human neuroglioma cells.16 However, we found that TFP at cytotoxic concentrations irreversibly disrupted lysosomal homeostasis especially in SCLC cells, driving LC3 conversion while blocking its degradation through autophagy. This was logical given that protonation of the weakly basic phenothiazines within lysosomes is expected to increase lumenal pH and could thereby adversely affect the activities of acid hydrolases.13, 17 Prolonged exposure to cytotoxic concentrations of TFP (10 μM in H82 and 20 μM in U-1810) was associated with lysosomal membrane permeabilization followed by mitochondrial depolarization. Accordingly, the protease inhibitor E-64d partially suppressed TFP-induced cell death. Moreover, we demonstrate that lysosomal targeting of TFP was critical for its cytotoxic activity because inhibition of vacuolar adenosine triphosphatase by BafA1, which dissipates the lysosomal pH gradient and prevents intra-lumenal entrapment of lysosomotropic compounds (e.g., phenothiazines) in their protonated forms,18 largely abolished TFP-induced cell death.
Overall, these data demonstrate that phenothiazines can modulate lysosomal functions in human LC cells, as the amount of endogenous LC3-II is correlated with autophagic vesicle formation and conversion of LC3-I into LC3-II is a hallmark of autophagic activity, and changes in these processes were observed upon phenothiazine treatment especially in SCLC.
The lysosome has recently emerged as a promising target for anticancer therapy because lysosome-initiated cell death can operate independently of p53 and downstream caspases,19, 20 pathways that are often functionally inactivated in a variety of human tumors.21, 22, 23 Also in our studies, lysosomal perturbation occurred despite mutation in p53 and was not influenced by intrinsic sensitivity/resistance toward conventional chemotherapeutic agents. From our data, we cannot rule out that SCLC cells or tumors with wt p53 or p53 null status could potentially respond differently, yet our opinion is that in this context, p53 status is not the main factor that regulates phenothiazine monotherapy response of SCLC. However, the disparate sensitivities that exist between, but also within, SCLC and NSCLC indicate intrinsic differences in lysosomal activity, suggesting that not all SCLC cells will be equally susceptible to phenothiazine-based treatment. Hence, we sought to reveal whether lysosome-associated parameters can be used to predict the sensitivity of human LC cells to phenothiazines. Indeed, using a limited number of SCLC and NSCLC cell lines, we found that sensitivity to phenothiazines inversely correlated with baseline LysoTracker Green retention, which was generally higher in NSCLC. Meanwhile, sensitivity to phenothiazines correlated positively with the magnitude by which LysoTracker retention was lost upon treatment, which tends to be more pronounced in SCLC. This suggests that lysosomes in the tested panel of SCLC cells may be less abundant and/or have higher intra-lumenal pH and are therefore more prone to functional perturbations. These findings require, however, further validation in a wider panel of SCLC cell lines to make a final conclusion. Also, as LysoTracker Green only stain lysosomes in live cells, alternative methodologies need to be developed to assess definitively whether lysosomal mass and/or pH is altered in clinical SCLC specimens such as, for example, tissue microarray staining with LC3-II upon treatment. Nevertheless, consistent with this idea, a number of different SCLC cell lines (H69, H82, H592 and U-1285) exhibited increased sensitivity toward several well-known lysosome-disrupting agents such as TMX, CQ, quinacrine and ammonium chloride (NH4Cl), all of which elicited larger changes in LysoTracker retention in SCLC cells as compared with NSCLC counterparts. By contrast, we found that the CaM antagonist W7 neither induced LC3-II accumulation nor was selectively cytotoxic in SCLC cells. In addition, the expression of D2R was comparable in our cell lines panel. Therefore, the enhanced cytotoxicity of phenothiazines in SCLC that we report here most likely derives from lysosomal dysregulation rather than preferential inhibition of CaM or neurotransmitter signaling, two established activities of phenothiazines.5 In addition, it is worth noting that NSCLC cell lines in general exhibited a higher basal autophagy than SCLC counterparts, which may render them less sensitive to chemically induced lysosomal perturbation. Similar observations have been reported by other investigators, implicating autophagy as a pro-survival mechanism in NSCLC.24, 25
Multiple studies on human and mouse cell lines have demonstrated the cytotoxic potential of phenothiazines given as monotherapy.4 In addition, a few case reports exist that described anecdotal evidence for the antitumor activity of phenothiazines in vivo.26, 27, 28 While the clinical utility of phenothiazines for anticancer treatment needs to be further analyzed through in vivo using SCLC xenograft, transgenic murine SCLC models and possibly also patient-derived xenografts, we believe our data in SCLC cell lines provide an important rationale for such analysis. Indeed, it has been shown that at advanced stages, several types of human cancers rely on lysosomes/autophagy for survival and metastasis.29 This is not surprising given that tumors frequently experience metabolic and replication stress.30, 31 Therefore, it is conceivable that tumors with high stress load and relatively low lysosomal activity could be particularly sensitive to phenothiazines. Given that SCLC initially are typically sensitive to conventional CT but rapidly develop resistance, our data support clinical trials where phenothiazines are administered as second-line treatment for tumors that become refractory to standard treatment. We therefore envision that the effects of phenothiazines per se as well as its potential chemosensitizing properties can help improve the outcome for relapsed SCLC patients who no longer respond to conventional treatment.
In summary, the present work uncovered a novel activity of phenothiazines as agents capable of inhibiting survival and inducing cell death in human SCLC (Figure 7), a tumor type that is notoriously difficult to treat with conventional CT because of its ability to rapidly acquire resistance. Moreover, we provide evidence showing that phenothiazine-induced disruption of lysosomal functions is critically required for phenothiazine-induced cell death effects, with SCLC especially susceptible because of their inherently low lysosomal number and/or pH. Finally, our data suggest that analysis of intrinsic lysosomal functions may identify SCLC cases that are most likely to respond to phenothiazine-based regimens, however, extended cell line analyses as well as in vivo studies are needed to formulate such conclusion. On the basis of these findings, we conclude that phenothiazines are suitable lead compounds for further development of lysosome-targeted treatment to combat SCLC and which merit further analysis for antitumor efficacy in patient-derived xenograft models and/or genetically modified mice models of SCLC.

A working model for the lysosome-dependent usage of phenothiazines in human LC. Chemical structure common to all phenothiazines followed by hypothetical mechanisms of actions in LC is presented. Lower lysosomal mass and buffer capacity of the SCLC cells results in perturbations of lysosomal functions causing cell death while NSCLC cells with higher lysosomal mass and buffer capacity survive
Materials and Methods
Cells lines and culture conditions
The following human LC cell lines were used: H69, H82, H592, U-1258, U-1568, U-1690, U-1906 and U-2020 (all SCLC); A549, H125, H1299, H157, H23, H661, U-1752 and U-1810 (all NSCLC). Cell lines whose name begin with the letter ‘U’ were established at the University of Uppsala, Sweden, while others were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) or Coriell Cell Repositories (Camden, NJ, USA). The characteristics of these cell lines as well as, for some of them, their p53 status is indicated in Supplementary Table S1. For comparison, we also used the non-cancerous cell line WI-38 (primary fetal lung fibroblasts).32 WI-38 was grown in MEM medium containing L-glutamine and supplemented with 15% fetal bovine serum (FBS). All other cell lines were maintained in RPMI-1640 medium containing L-glutamine and supplemented with 10% FBS.
Chemicals and reagents
TFP, FPZ, CPZ, TFPZ, PZ, (all from Sigma-Aldrich, Stockholm, Sweden) and cis-FPX (H Lundbeck A/S, Copenhangen, Denmark) were prepared in dimethyl sulfoxide and used at a final concentration of 0–20 μM. TMX, CQ diphosphate, BafA1, NH4Cl, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride (W7), L-741626, acridine orange, fluorescein di-β-D-galactopyranoside (FDG) and carboxyfluorescein diacetate N-succinimidyl ester (CFSE) were from Sigma-Aldrich. z-VAD-fmk, E-64d and CHX were from BD PharMingen (San Jose, CA, USA), Enzo Life Sciences (Ann Arbor, MI, USA) and Sigma-Aldrich, respectively. PI, tetramethylrhodamine ethyl ester perchlorate (TMRE) and LysoTracker Green DND-26 were from Invitrogen, Stockholm, Sweden.
Cell viability analysis
Cell viability was analyzed with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assay as previously described.33 Briefly, 5000–10 000 cells per well were seeded in 96-well plates. Twenty-four hours after seeding, cells were subjected to a concentration range of phenothiazines alone as indicated in the figures, or were treated with cisplatin, gemcitabine or etoposide. Cell viability was determined after 72 h of continuous treatment where the viability of untreated cells was arbitrary set to 1. Alternatively, area under the curve was calculated in the GraphPad Prism software (GraphPad Prism Software Inc., La Jolla, CA, USA). Cytotoxicity values reported in Table 1 and Supplementary Figure S1 were calculated by the following formula: cytotoxicity=100%−viability (%).
Flow cytometry
Fluorescent probe-based analyses of intracellular organelles were performed in non-fixed cells. Labeling of lysosomes was performed using LysoTracker Green DND-26 (50 nM, 1 h). Lysosome-associated β-gal activity was assayed by enzymatic cleavage of FDG (50 μM, 30 min). Mitochondrial transmembrane potential was determined by retention of the potentiometric dye TMRE (50 nM, 30 min). Cellular proliferative capacity was assessed using the CFSE-labeling method, as previously described.34 DNA content analysis was performed in ethanol-fixed cells by PI staining. At least 10 000 events were recorded on a Becton-Dickinson FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Data analysis was conducted using the built-in Cell Quest software of FACSCalibur flow cytometer (BD Biosciences). Cell cycle distribution was assigned using ModFit (Verity Software House, Topsham, ME, USA).
Microscopy
Gross morphology of cells was examined under a Nikon Eclipse TS100 light microscope (Nikon Instruments Europe B.V., Amsterdam, The Netherlands) at × 10 magnification.
Immunoblotting
Whole-cell lysate (WCL) was obtained by lysing cells in radio-immunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.1% Na-deoxycholate, 1% NP-40) supplemented with protease and phosphatase inhibitor cocktail tablets (Roche Diagnostics AB, Stockholm, Sweden). Thirty micrograms of WCL were resolved by SDS-PAGE (NuPAGE, Invitrogen). Immunoblotting was performed with primary antibodies recognizing PARP, LAMP-1 (both Santa Cruz Biotechnology, Santa Cruz, CA, USA), LC3B (Cell Signaling Technology, Danvers, MA, USA), p62 (BD Transduction Laboratories, San Jose, CA, USA), D2R (Abcam, Cambridge, UK) and GAPDH (Trevigen, Gaithersburg, MD, USA). IR Dye-linked secondary antibodies (LI-COR Biosciences, Bad Homburg, Germany) were used to image bands on the Odyssey Sa Infrared Imaging System (LI-COR).
Statistical analysis
Unless otherwise stated, all experiments were repeated three times. Data points are expressed as the mean±S.D. Statistical significance was tested by two-tailed unpaired Student’s t-test. Asterisk indicates P<0.05.
Abbreviations
- BafA1:
-
bafilomycin A1
- CaM:
-
calmodulin
- CFSE:
-
carboxyfluorescein diacetate N-succinimidyl ester
- CHX:
-
cycloheximide
- cis-FPX:
-
cis-flupenthixol dihydrochloride
- CNS:
-
central nervous system
- CPZ:
-
chlorpromazine hydrochloride
- CQ:
-
chloroquine
- CT:
-
chemotherapy
- D2R:
-
dopamine D2 receptor
- FBS:
-
fetal bovine serum
- FDG:
-
fluorescein di-β-D-galactopyranoside
- FPZ:
-
fluphenazine dihydrochloride
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- LAMP-1:
-
lysosomal-associated membrane protein 1
- LC:
-
lung cancer
- LC3:
-
light chain 3
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- NH4Cl:
-
ammonium chloride
- NSCLC:
-
non-small cell lung carcinoma
- PARP:
-
poly (ADP-ribose) polymerase
- PI:
-
propidium iodide
- PZ:
-
promazine hydrochloride
- SCLC:
-
small cell lung carcinoma
- TFP:
-
trifluoperazine dihydrochloride
- TFPZ:
-
triflupromazine hydrochloride
- TMRE:
-
tetramethylrhodamine ethyl ester perchlorate
- TMX:
-
tamoxifen
- WCL:
-
whole-cell lysate
- z-VAD-fmk:
-
benzyloxycarbonyl-Val-Ala-Asp fluoromethylketone
- β-gal:
-
β-galactosidase
References
Shen WW . A history of antipsychotic drug development. Compr Psychiatry 1999; 40: 407–414.
Roth BL, Sheffler DJ, Kroeze WK . Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 2004; 3: 353–359.
Sudeshna G, Parimal K . Multiple non-psychiatric effects of phenothiazines: a review. Eur J Pharmacol 2010; 648: 6–14.
Motohashi N, Kawase M, Satoh K, Sakagami H . Cytotoxic potential of phenothiazines. Curr Drug Targets 2006; 7: 1055–1066.
Hait WN, Lee GL . Characteristics of the cytotoxic effects of the phenothiazine class of calmodulin antagonists. Biochem Pharmacol 1985; 34: 3973–3978.
Drori S, Eytan GD, Assaraf YG . Potentiation of anticancer-drug cytotoxicity by multidrug-resistance chemosensitizers involves alterations in membrane fluidity leading to increased membrane permeability. Eur J Biochem 1995; 228: 1020–1029.
Lee MS, Johansen L, Zhang Y, Wilson A, Keegan M, Avery W et al. The novel combination of chlorpromazine and pentamidine exerts synergistic antiproliferative effects through dual mitotic action. Cancer Res 2007; 67: 11359–11367.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D . Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90.
Nordenberg J, Fenig E, Landau M, Weizman R, Weizman A . Effects of psychotropic drugs on cell proliferation and differentiation. Biochem Pharmacol 1999; 58: 1229–1236.
Sridhar KS, Krishan A, Samy TS, Sauerteig A, Wellham LL, McPhee G et al. Prochlorperazine as a doxorubicin-efflux blocker: phase I clinical and pharmacokinetics studies. Cancer Chemother Pharmacol 1993; 31: 423–430.
Rossi A, Martelli O, Di Maio M . Treatment of patients with small-cell lung cancer: from meta-analyses to clinical practice. Cancer Treat Rev 2012; 39: 498–506.
Daniel WA . Mechanisms of cellular distribution of psychotropic drugs. Significance for drug action and interactions. Prog Neuropsychopharmacol Biol Psychiatry 2003; 27: 65–73.
Kaufmann AM, Krise JP . Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications. J Pharm Sci 2007; 96: 729–746.
Hann CL, Rudin CM . Management of small-cell lung cancer: incremental changes but hope for the future. Oncology (Williston Park) 2008; 22: 1486–1492.
Hait WN, Morris S, Lazo JS, Figlin RJ, Durivage HJ, White K et al. Phase I trial of combined therapy with bleomycin and the calmodulin antagonist, trifluoperazine. Cancer Chemother Pharmacol 1989; 23: 358–362.
Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA 2007; 104: 19023–19028.
Poole B, Ohkuma S . Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol 1981; 90: 665–669.
Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol 2006; 69: 1125–1136.
Broker LE, Kruyt FA, Giaccone G . Cell death independent of caspases: a review. Clin Cancer Res 2005; 11: 3155–3162.
Erdal H, Berndtsson M, Castro J, Brunk U, Shoshan MC, Linder S . Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis. Proc Natl Acad Sci USA 2005; 102: 192–197.
Galluzzi L, Vitale I, Vacchelli E, Kroemer G . Cell death signaling and anticancer therapy. Front Oncol 2011; 1: 5.
Joseph B, Lewensohn R, Zhivotovsky B . Role of apoptosis in the response of lung carcinomas to anti-cancer treatment. Ann N Y Acad Sci 2000; 926: 204–216.
Rodriguez-Nieto S, Zhivotovsky B . Role of alterations in the apoptotic machinery in sensitivity of cancer cells to treatment. Curr Pharm Des 2006; 12: 4411–4425.
Kaminskyy VO, Piskunova T, Zborovskaya IB, Tchevkina EM, Zhivotovsky B . Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012; 8: 1032–1044.
Karpathiou G, Sivridis E, Koukourakis MI, Mikroulis D, Bouros D, Froudarakis ME et al. Light-chain 3A autophagic activity and prognostic significance in non-small cell lung carcinomas. Chest 2011; 140: 127–134.
Csatary LK . Chlorpromazines and cancer. Lancet 1972; 2: 338–339.
Hercbergs A . Thioridazine: a radiation enhancer in advanced cervical cancer? Lancet 1988; 2: 737.
Jones GR . Successful cancer therapy with promethazine: the rationale. Med Hypotheses 1996; 46: 25–29.
Castino R, Demoz M, Isidoro C . Destination 'lysosome': a target organelle for tumour cell killing? J Mol Recognit 2003; 16: 337–348.
Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21: 1621–1635.
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21: 1367–1381.
Hayflick L, Moorhead PS . The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621.
Abate G, Mshana RN, Miorner H . Evaluation of a colorimetric assay based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) for rapid detection of rifampicin resistance in Mycobacterium tuberculosis. Int J Tuberc Lung Dis 1998; 2: 1011–1016.
Zong D, Haag P, Yakymovych I, Lewensohn R, Viktorsson K . Chemosensitization by phenothiazines in human lung cancer cells: impaired resolution of gammaH2AX and increased oxidative stress elicit apoptosis associated with lysosomal expansion and intense vacuolation. Cell Death Dis 2011; 2: e181.
Acknowledgements
This research was supported by grants from the Swedish Cancer Society, the Stockholm Cancer Society, the Stockholm County Council, the Swedish Research Foundation, Swedish Board of National Health and Welfare, the Karolinska Institutet and the European Union (EC FP-6 Chemores and FP-7 Apo-Sys).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Ciliberto
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zong, D., Zielinska-Chomej, K., Juntti, T. et al. Harnessing the lysosome-dependent antitumor activity of phenothiazines in human small cell lung cancer. Cell Death Dis 5, e1111 (2014). https://doi.org/10.1038/cddis.2014.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.56
Keywords
This article is cited by
-
Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles
Cell Research (2016)
-
Thioridazine inhibits gene expression control of the cell wall signaling pathway (CWI) in the human pathogenic fungus Paracoccidioides brasiliensis
Molecular Genetics and Genomics (2016)
