
Abstract
The interleukin-1 (IL-1) family of cytokines has been implicated in the pathogenesis of atherosclerosis in previous studies. The NLRP3 inflammasome has recently emerged as a pivotal regulator of IL-1β maturation and secretion by macrophages. Little is currently known about a possible role for the NLRP3 inflammasome in atherosclerosis progression in vivo. We generated ApoE−/− Nlrp3−/−, ApoE−/− Asc−/− and ApoE−/− caspase-1−/− double-deficient mice, fed them a high-fat diet for 11 weeks and subsequently assessed atherosclerosis progression and plaque phenotype. No differences in atherosclerosis progression, infiltration of plaques by macrophages, nor plaque stability and phenotype across the genotypes studied were found. Our results demonstrate that the NLRP3 inflammasome is not critically implicated in atherosclerosis progression in the ApoE mouse model.
Similar content being viewed by others

Main
Atherosclerosis is implicated in 50% of deaths in developed countries, and is thus a major health issue. Although the precise pathogenesis of the disease is complex and remains incompletely understood, it is generally agreed that inflammation has a pivotal role.1 In particular, the release of proinflammatory cytokines by atherosclerotic plaque-infiltrating macrophages is seen as critical. Interleukin-1β (IL-1β) is produced by activated macrophages and considered a master switch of inflammation. IL-1β and its related parent IL-1α have both been implicated in the pathogenesis of atherosclerosis in previous studies.2
Recent advances have shed light on the molecular mechanisms governing the regulation of IL-1β maturation and secretion by macrophages. NOD-like receptors (NLRs) are a family of cytoplasmic pattern-recognition receptors that have critical roles in innate immunity.3 In particular, some NLRs have been shown to form caspase-1-activating cytoplasmic complexes called inflammasomes, responsible for the processing of pro-IL-1β into its mature active form and its subsequent secretion.4 The NLRP3 inflammasome is assembled around the NLR scaffold protein NLRP3 upon detection of pathogen-associated molecular patterns or endogenous danger signals (danger-associated molecular patterns). Upon activation, NLRP3 recruits the adapter protein ASC, which in turn recruits procaspase-1.4 When brought into close proximity, procaspase-1 molecules are able to transactivate each other, resulting in the processing of pro-IL-1β.
The NLRP3 inflammasome has attracted considerable attention because of its implication in several human diseases and the availability of inhibitors of IL-1β that have been recently very successful in clinical trials with patients suffering from gout and type 2 diabetes.5 We therefore addressed the issue of a possible role for the NLRP3 inflammasome in atherosclerosis development.
Results
One of the reasons proposed to lead to the development of atherosclerosis is the presence of oxidized low-density lipoproteins (oxLDLs).6 Indeed, we found that cholesterol oxidative derivatives such as 7-ketocholesterol, a major component of oxLDL detectable in atherosclerotic plaques,6 induces secretion of the processed form of caspase-1 by murine primary macrophages in an NLRP3 inflammasome-dependent manner in vitro (Figure 1a).

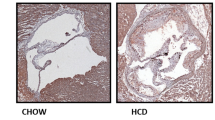
NLRP3 inflammasome deficiency does not impair atherosclerosis progression in ApoE−/− mice in vivo. (a) Lipopolysaccharide-primed murine bone marrow-derived macrophages isolated from Nlrp3+/+ and Nlrp3−/− mice were stimulated with 7-ketocholesterol and uric acid crystals (MSU, 150 μg/ml) for 8 h as indicated and analyzed by western blot. Active caspase-1 (mCasp1) released into the supernatant (SN) was measured by western blot to determine NLRP3 inflammasome activation. (b) Oil Red O staining of thoracoabdominal aorta and MOVATS staining of aortic valve plaques across the four indicated genotypes. Data are shown as mean±S.E.M. The asterisk denotes P<0.05
The ApoE−/− mouse develops severe hypercholesterolemia and spontaneous atherosclerosis, and is therefore a widely accepted experimental mouse model to study atherosclerosis.7 Considering the capacity of 7-ketocholesterol to activate the NLRP3 inflammasome, we next wanted to determine a possible role of the NLRP3 inflammasome in atherosclerosis development in vivo. To this end, we crossed Nlrp3−/−, Asc−/− or caspase-1−/− mice with ApoE−/− mice. Contrary to our expectations, we found little differences between double knockouts and controls with respect to atherosclerotic plaque surface across the aorta (Figure 1b) or the aortic valve (Figure 1b) of these mice following 11 weeks on a high-fat diet. Cholesterol crystals were similarly abundant in plaques of all mice. Accordingly, plaque macrophage infiltration was equivalent in all mice, as determined by Mac-2 staining (Figure 2), suggesting that NLRP3 inflammasome-deficient macrophages are equally recruited in atherosclerotic plaques.

Equal recruitment of macrophages to atherosclerotic plaques in NLRP3 inflammasome-deficient ApoE−/− mice. Plaque-infiltrating macrophages were assessed with an anti-Mac-2 staining. Data are shown as mean±S.E.M.
In patients suffering from atherosclerosis, over 60% of myocardial infarctions are caused by low-grade (<50%) coronary artery stenosis.8 Therefore, the sheer surface of atherosclerotic plaques is clinically less crucial than their stability. Plaque smooth muscle cell content, a marker of plaque stability, was quantified by anti-α-smooth muscle actin staining (Figure 3). Again, we found no significant differences between the four genotypes. Furthermore, plaque phenotype was similar in all mice, showing lesions in advanced stage (Table 1). Although ApoE−/− ASC−/− mice featured significantly lower levels of plaque layering and adventitial inflammation, this was offset by an increased prevalence of thinned fibrous cap, suggesting that, overall, the plaque phenotype was not decisively more or less favorable than controls (Table 1).

Plaque stability is unaltered in NLRP3 inflammasome-deficient ApoE−/− mice. Smooth muscle actin (SMA) staining of atherosclerotic plaques across the four indicated genotypes
Taken together, we conclude that NLRP3 inflammasome deficiency does not affect atherosclerosis progression or plaque phenotype in ApoE−/− mice.
Discussion
Atherosclerosis is increasingly seen as a chronic inflammatory disease of the vessel wall. In this context, plaque-infiltrating macrophages and the proinflammatory cytokines that they secrete have critical roles. IL-1β is a master switch of inflammation and has thus attracted considerable attention regarding a putative role in the pathogenesis of atherosclerosis.
In the present study, we show that the absence of the NLRP3 inflammasome key components NLRP3, ASC or caspase-1 does not influence atherosclerosis progression, infiltration of plaques by macrophages, nor plaque stability in an ApoE mouse model.
Our results are at odds with a recent report by Latz and coworkers.9 Although our data agree that molecules reportedly associated with atherosclerosis, such as cholesterol crystals or oxidized lipids, can potently activate the NLRP3 inflammasome in vitro, our results concerning the involvement of the NLRP3 inflammasome in atherosclerosis development in vivo differ. Unlike in the present study, these authors used mice deficient in the low-density lipoprotein receptor that were transplanted with Nlrp3-, Asc- or IL-1α/β-deficient bone marrow cells after lethal irradiation as a model of atherosclerosis. Compared with cells from wild-type mice, they observed a 60% decrease in total lesion size at the aortic sinus upon transfer of NLRP3-, ASC- or IL-1α/β-deficient cells, and thus proposed an implication of the NLRP3 inflammasome in disease progression. Unfortunately, they did not assess plaque quality or stability.
What could account for the divergence observed between these two studies? One simple explanation may be the difference in the mouse model used to study atherosclerosis. Although both the ApoE−/− and Ldlr−/− mouse strains are known to efficiently develop atherosclerotic lesions, some notable differences exist. First, ApoE−/− mice are generally more hypercholesterolemic than Ldlr−/− mice on a similar high-fat diet; therefore, minor contributions of genes possibly contributing to atherosclerosis might be missed in the former model. Moreover, using the bone marrow transplantation model, it is only possible to assess the contribution of bone marrow-derived cells, but other cells can also release active IL-1β and so other cellular sources of IL-1β may influence disease progression. However, under such a hypothesis, one would expect differences between genotypes to be more pronounced in our double-knockout mice.
Another explanation for the discrepancy could be that mostly IL-1α and not IL-1β contributes to the pathogenesis of atherosclerosis in ApoE−/− mice. Indeed, as active IL-1α can be generated in the absence of the NLRP3 inflammasome via a calpain-mediated processing,10 the absence of the NLRP3 inflammasome would not impair the progression of atherosclerosis. Moreover, IL-1α, like IL-1β, signals through the IL-1 receptor. This hypothesis is supported by several studies, which primarily investigated the role of the IL-1R and its naturally-occurring endogenous antagonist (IL-1Ra) in the pathogenesis of atherosclerosis.2 IL-1R-deficient ApoE−/− mice were shown to feature decreased atherosclerosis.11 In accordance, genetically induced overexpression of IL-1Ra (which blocks receptor access of both IL-1α and IL-1β) or infusion of its recombinant form in ApoE−/− mice resulted in decreased atherosclerosis.12, 13 Conversely, IL-1Ra-deficient mice displayed enhanced atherosclerosis.14, 15
IL-1α deficiency was reported to be associated with a 50% decrease in atherosclerotic plaque area using a model of C57BL/6 mice fed a high-fat diet.16 In that study, IL-1β deficiency did not impair atherosclerosis progression in a statistically significant manner. In another study, aortic sinus lesion area in IL-1α-deficient ApoE−/− mice was lower than in IL-1β-deficient ApoE−/− mice.17
Taken together, our results suggest that atherogenesis in ApoE−/− mice can progress independently of the NLRP3 inflammasome, a complex that appears to be crucial in a different mouse model of atherosclerosis. The most reasonable explanations of the observed discrepancy between the two studies lie in the different mouse models used, coupled to a putative role of IL-1α in atherosclerosis development in the ApoE mouse model. Clearly, further research is warranted regarding the implication of inflammasomes and specific IL-1 family members in the pathogenesis of atherosclerosis.
Materials and Methods
Bone marrow-derived macrophages preparation, stimulation and western blot
Primary murine macrophages were obtained from tibial and femoral bone marrow cells as described elsewhere.18 Cells were primed overnight with 100 ng/ml ultra-pure lipopolysaccharide (Invivogen, San Diego, CA, USA), and stimulated the following day in OPTIMEM for 8 h. Uric acid and 7-ketocholesterol were purchased from Sigma (Buchs, Switzerland). Cells extracts and precipitated supernatants were analyzed by western blot. The antibody against mouse caspase-1 (p20) is a generous gift from Dr. Peter Vandenabeele (Ghent University).
Mice and atherosclerosis progression determination
ApoE−/− mice were from Jackson Laboratories (Bar Harbor, ME, USA; no. 002052); Nlpr3−/−,19 ASC−/−20 and caspase-1−/−21 mice have been described. All mice were on C57BL/6 background. ApoE−/− mice were first crossed with Nlrp3−/−, ASC−/− or caspase-1−/−, and the resulting F1 mice were then backcrossed on the ApoE−/− background. F2 mice were then crossed again to obtain double-deficient mice. The number of mice studied for each genotype was as follows: ApoE−/−, n=17; ApoE−/− Nlrp3−/−, n=18; ApoE−/− caspase-1−/−, n=13; ApoE−/− ASC−/−, n=11. The 4-week-old mice were fed a high-fat diet (KlibaNafag 3200 supplemented with 1.25% w/w cholesterol and 15% w/w cacao butter; Kliba Nafag, Kaiseraugst, Switzerland) for 11 weeks and subsequently killed. ApoE−/− mice fed a high-fat diet were used as controls. Detailed procedures of plaque surface, macrophage and smooth muscle cell quantification, as well as plaque quality assessment are described elsewhere.22
Statistical analysis
Data are shown as mean±S.E.M. Statistical analysis was carried out using a one-way analysis of variance coupled to a Dunnett post hoc test, except for the assessment of plaque quality that relied on Fisher's exact test. Statistical significance was defined as P<0.05.
Abbreviations
- NLR:
-
Nod-like receptors
- PRR:
-
pattern recognition receptors
- DAMP:
-
danger-associated molecular patterns
References
Hansson GK, Libby P . The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol 2006; 6: 508–519.
Kleemann R, Zadelaar S, Kooistra T . Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res 2008; 79: 360–376.
Martinon F, Mayor A, Tschopp J . The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27: 229–265.
Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Dinarello CA . Anti-inflammatory agents: present and future. Cell 2010; 140: 935–950.
Brown AJ, Jessup W . Oxysterols and atherosclerosis. Atherosclerosis 1999; 142: 1–28.
Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 2007; 27: 1706–1721.
Schroeder AP, Falk E . Vulnerable and dangerous coronary plaques. Atherosclerosis 1995; 118 (Suppl): S141–S149.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464: 1357–1361.
Carruth LM, Demczuk S, Mizel SB . Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J Biol Chem 1991; 266: 12162–12167.
Chi H, Messas E, Levine RA, Graves DT, Amar S . Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmacotherapeutic implications. Circulation 2004; 110: 1678–1685.
Devlin CM, Kuriakose G, Hirsch E, Tabas I . Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci USA 2002; 99: 6280–6285.
Merhi-Soussi F, Kwak BR, Magne D, Chadjichristos C, Berti M, Pelli G et al. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc Res 2005; 66: 583–593.
Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M et al. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2004; 24: 1068–1073.
Isoda K, Ohsuzu F . The effect of interleukin-1 receptor antagonist on arteries and cholesterol metabolism. J Atheroscler Thromb 2006; 13: 21–30.
Kamari Y, Werman-Venkert R, Shaish A, Werman A, Harari A, Gonen A et al. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 2007; 195: 31–38.
Kamari Y, Shaish A, Shemesh S, Vax E, Grosskopf I, Dotan S et al. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1α. Biochem Biophys Res Commun 2011; 405: 197–203.
Didierlaurent A, Brissoni B, Velin D, Aebi N, Tardivel A, Käslin E et al. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol 2006; 26: 735–742.
Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J . Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–241.
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004; 430: 213–218.
Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 1995; 267: 2000–2003.
Mazzolai L, Duchosal MA, Korber M, Bouzourene K, Aubert JF, Hao H et al. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE-/- mice. Hypertension 2004; 44: 277–282.
Acknowledgements
This work was supported by the Swiss National Science Foundation through an MD-PhD grant (to PM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Menu, P., Pellegrin, M., Aubert, JF. et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis 2, e137 (2011). https://doi.org/10.1038/cddis.2011.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.18
Keywords
This article is cited by
-
Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis
Cell Death & Disease (2022)
-
Oxidative Stress-Mediated Programmed Cell Death: a Potential Therapy Target for Atherosclerosis
Cardiovascular Drugs and Therapy (2022)
-
Anti-inflammatory Action of Statins in Cardiovascular Disease: the Role of Inflammasome and Toll-Like Receptor Pathways
Clinical Reviews in Allergy & Immunology (2021)
-
Artemisinin suppresses myocardial ischemia–reperfusion injury via NLRP3 inflammasome mechanism
Molecular and Cellular Biochemistry (2020)
-
Diversity of macrophage phenotypes and responses in atherosclerosis
Cellular and Molecular Life Sciences (2020)
