
Abstract
Diabetes is associated with the death and dysfunction of insulin-producing pancreatic β-cells. In other systems, Musashi genes regulate cell fate via Notch signaling, which we recently showed regulates β-cell survival. Here we show for the first time that human and mouse adult islet cells express mRNA and protein of both Musashi isoforms, as well Numb/Notch/Hes/neurogenin-3 pathway components. Musashi expression was observed in insulin/glucagon double-positive cells during human fetal development and increased during directed differentiation of human embryonic stem cells (hESCs) to the pancreatic lineage. De-differentiation of β-cells with activin A increased Msi1 expression. Endoplasmic reticulum (ER) stress increased Msi2 and Hes1, while it decreased Ins1 and Ins2 expression, revealing a molecular link between ER stress and β-cell dedifferentiation in type 2 diabetes. These effects were independent of changes in Numb protein levels and Notch activation. Overexpression of MSI1 was sufficient to increase Hes1, stimulate proliferation, inhibit apoptosis and reduce insulin expression, whereas Msi1 knockdown had the converse effects on proliferation and insulin expression. Overexpression of MSI2 resulted in a decrease in MSI1 expression. Taken together, these results demonstrate overlapping, but distinct roles for Musashi-1 and Musashi-2 in the control of insulin expression and β-cell proliferation. Our data also suggest that Musashi is a novel link between ER stress and the compensatory β-cell proliferation and the loss of β-cell gene expression seen in specific phases of the progression to type 2 diabetes.
Similar content being viewed by others


Main
The destruction and dysfunction of pancreatic insulin-producing β-cells are hallmarks of diabetes, but the molecular links between the stresses that cause β-cell death and pathways that control β-cell function and differentiation are unclear. Although once thought to be quiescent and terminally differentiated, we and others have shown that individual adult pancreatic cells exhibit a dynamic range in differentiation status.1, 2, 3 Indeed, accumulating evidence reveals striking plasticity between pancreatic cell types and the islet endocrine lineages,4, 5 but the effects of cellular stress on plasticity pathways remain unknown.
Notch and neurogenin-3 (Ngn3) have established roles in pancreas development.6 Ngn3 is essential for the development of all pancreatic endocrine lineages.6 During embryonic development, Notch negatively regulates Ngn3 via its target gene Hes1.7 Thus, the Notch signaling pathway is critical at maintaining a proliferating pool of endocrine progenitor cells and influences the timely cell lineage specification of exocrine and endocrine cell differentiation.8 Accumulating evidence suggests that these developmental gene networks also have roles in the adult. We have demonstrated that Ngn3 mRNA and protein are present in adult human and mouse islets.9 Inhibiting Notch signaling with N-[(3,5-difluorophenyl)acetyl]-L-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester (DAPT) increased Ngn3 levels and induced apoptosis, suggesting an important role for this pathway in adult β-cell survival.9 Subsequently, it was shown that conditional knockout of Ngn3 in adult islets impaired β-cell function.10 Unfortunately, little is known about the factors that regulate Notch and Ngn3 signaling in the adult pancreatic β-cell.
In other tissues, the RNA-binding protein Musashi, a translational repressor of Numb,11 regulates cell fate via Notch signaling by maintaining a pool of self-renewing stem cells.12, 13 Targeted disruption of Musashi-1 (Msi1) and Musashi-2 (Msi2) revealed their roles in maintaining neural stem cell populations in the adult central nervous system.14 Expression of Msi1 and Hes1 was also reported in the stem cell niche of highly proliferative intestinal crypt cells.15 The Musashi target Numb plays a role in determining cell fate during embryonic development in some tissues16 and can also be found in the developing mouse pancreas.17 To date, there are no reports regarding Musashi expression in developing or mature β-cells or its possible mechanistic roles. We hypothesized that Musashi expression remains in a subset of adult β-cells and that it regulates cell fate decisions in response to cellular stress.
Here, we show that both Musashi isoforms are expressed in developing and adult human and mouse islets and β-cells at the transcript and protein levels. Musashi was more abundant in dedifferentiated and immature β-cells. Conditions associated with type 2 diabetes, such as endoplasmic reticulum (ER) stress induced by sarco-endoplasmic reticulum Ca2+ ATPase inhibition (ER Ca2+ depletion) or palmitate treatment, increased the expression of the Musashi genes. Overexpression of MSI1 in mouse insulinoma cell line 6 (MIN6) β-cells increased Hes1 gene expression, increased proliferation and downregulated insulin gene expression, whereas knockdown had the opposite effects. Our data suggest that Musashi genes regulate insulin expression, apoptosis and proliferation in response to ER stress via Hes1, but independently of the Numb/Notch pathway.
Results
Musashi is expressed in adult pancreatic islet cells
The expression and function of Notch pathway components in adult islets was recently demonstrated,9, 10 but the upstream regulators of Notch are unclear in these cells. Our identification of MSI1 via mass spectroscopy in a proteomics study of human islets provided the impetus to investigate this translational repressor protein. Published gene expression databases, Tag-Seq libraries and microarray data sets of fluorescence-activated cell sorting (FACS)-purified β-cells18, 19 pointed to the presence of both Musashi homologs in adult islets and purified β-cells from humans, mice and rats, as well as Numb/Numb-like, Notch1-4, Hes1 and Ngn3 (Figure 1a). However, the functional relationships between the components of this classical Notch pathway remain understudied in adult β-cells.

Expression of Musashi genes in pancreatic islets and β-cells. (a) mRNA expression data were extracted from four independent sources for Musashi-1 (Msi1) and Musashi-2 (Msi2), as well as other Notch pathway genes for human, mouse and/or rat islets and β-cells . Expression levels for T1DBase (relative expression), FACS-purified β-cells and purified islets microarray (relative expression), MPSS (tpm=transcripts per million) and Tag-Seq library (tpm=tags per million) are displayed by color (see look-up table; nd, data not determined). (b) RT-PCR analysis of human Msi1 (using two different primer sets spanning different exons) and Msi2 in human islets (representative of >3 experiments). (c) RT-PCR analysis of Msi1 and Msi2 (using two different primer sets spanning different exons) in mouse islets. (d) RT-qPCR expression analysis of Msi1 and Msi2 in mouse islets (n=4) and pituitary gland (n=3). Glucagon and insulin staining in adult human pancreas along with Msi1 (e) and Msi2 (f). Scale bar=100 μm. Glucagon and insulin staining in adult mouse pancreas along with Msi1 (g) and Msi2 (h). Scale bar=20 μm. (i) Deconvolution microscopy of Msi1 and Msi2 subcellular location in MIN6 cells. Scale bar=5 μm. Similar results were observed with overexpression of Msi1 and Msi2 fluorescent fusion proteins. (j) Western blot for Msi1 protein in MIN6 cells, islets and exocrine tissue from mice aged 6, 12 and 30 weeks (n=3). Mouse brain tissue was used as a positive control. *P<0.05
We next examined the expression of Msi1 and Msi2 at the mRNA and protein levels in samples of human and mouse islets. Reverse transcriptase-polymerase chain reaction (RT-PCR) analysis showed that both homologs of Musashi were expressed in adult human and mouse islets (Figures 1b and c). Reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) showed that the expression level of Msi2 was several fold higher compared with Msi1 in mouse islets (Figure 1d), consistent with the results obtained from gene expression databases (Figure 1a). The levels of Msi1 and Msi2 in the islets were similar to those found in the mouse pituitary (Figure 1d). Immunofluorescence staining confirmed the presence of MSI1 and MSI2 in a subset of human glucagon-positive α-cells and insulin-positive β-cells (Figures 1e and f). MSI1 was found in 61.9±30.1% of β-cells, while 72.5±3.3% of β-cells were positive for MSI2. Similarly, the percentages of MSI1- and MSI2-positive α-cells were 76.2±21.1% and 85.3±0.04%, respectively. Similar observations were made in mouse α- and β-cells (Figures 1g and h). We examined the subcellular location of endogenous Msi1 and Msi2 in MIN6 cells using deconvolution microscopy and found a punctate cytoplasmic localization consistent with a regulation of translation (Figure 1i). Interestingly, Msi1 protein levels increased with age in adult mice (Figure 1j), pointing to the possibility that stress associated with normal aging can upregulate Musashi levels. Overall, these results confirm the expression of Musashi genes at the mRNA and protein levels in adult human and mouse islets and β-cells, as well as in β-cell lines.
Musashi expression dynamics in human development and hESC differentiation
As Musashi has been proposed as a stem cell or progenitor cell marker regulating cell fate during differentiation,20 we probed for its expression in human fetal pancreas and during directed differentiation of human embryonic stem cells (hESCs) towards the pancreatic lineage (Figures 2 and 3). Both MSI1 and MSI2 proteins were present in developing human fetal pancreas at 8 weeks, co-staining with insulin (MSI1, 44.9%; MSI2, 56.1%) and glucagon (MSI1, 50.1%; MSI2, 81.8%). At 12 weeks, the percentage of insulin-positive cells with MSI1 or MSI2 was 67.3% and 79.3%, respectively, whereas the percentage of glucagon-positive cells with MSI1 or MSI2 was 72.2% and 95.9%, respectively (Figures 2a–d). As a positive control, we observed both Musashi proteins in human fetal intestine (Figures 2e and f). Notably, although many of the cells were Musashi-positive, staining for both Musashi proteins was stronger in glucagon-positive or insulin/glucagon double-positive cells compared to cells positive only for insulin (Figures 2a-d). Recent analysis of human fetal pancreas and hESC differentiation has demonstrated that insulin/glucagon double-positive cells are common and may be the progenitors of mature α-cells.21, 22

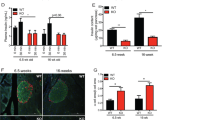
Expression of MSI1 and MSI2 during human fetal development. (a–d) Glucagon and insulin staining in 8- and 12-week human fetal pancreatic sections along with MSI1 (a and c) and MSI2 (b and d) staining. Scale bar=100 and 20 μm for merged and expanded images, respectively. (e) MSI1 and (f) MSI2 staining in 12-week human fetal intestine sections. Scale bar=200 μm

Increased expression of Msi1 and Msi2 during human stem cell differentiation towards an endocrine fate. (a) RT-qPCR analysis of Msi1 and Msi2 mRNA levels during the differentiation of two independent hESC lines into definitive endoderm (n=3). *P<0.05 DE versus hESCs for respective gene. (b) Msi1, Msi2 and Pdx1 mRNA expression during the 28-day differentiation culture of hESCs towards an endocrine fate (n=3). Human islets are shown for comparison. *P<0.05 relative to day 1 (undifferentiated hESCs) for respective gene; #P<0.05 relative to day 4 (definitive endoderm) for respective gene; ND=not detected. (c) Images of glucagon, insulin, MSI1 and MSI2 immunoreactivities in differentiated hESCs at day 28 of culture. Scale bar=50 μm
We next sought to determine if Musashi expression changed during hESC differentiation towards the pancreatic lineage. The mRNA expression levels of both MSI1 and MSI2 increased significantly during the course of hESC differentiation from undifferentiated cells to definitive endoderm and subsequently to PDX1-positive developing pancreatic progenitor cells (Figures 3a and b). These changes in Musashi expression were consistent between two different hESC lines (Figure 3a). MSI1 and MSI2 mRNA levels in adult human islets were similar to hESC-derived cultures on days 11 and 14, whereas PDX1 mRNA levels were significantly higher in adult human islets compared with all stages of hESC differentiation (Figure 3b). Both Musashi proteins were also present in differentiated hESC (day 28), a stage where pancreatic and endocrine progenitors, immature endocrine cells and mature endocrine cells are all present, and colocalizing with a subset of insulin-positive cells (MSI1, 61.4%; MSI2, 67.9%) and glucagon-positive cells (MSI1, 45.6%; MSI2, 68.9%) (Figure 3c). Collectively, these data suggest a role for Musashi proteins in the fetal pancreas and directed progenitor cell differentiation, but cast doubt that they represent true stem cell factors in this system.
Increased Musashi expression with decreased β-cell differentiation status
We have previously shown that single adult β-cells from humans, mice and β-cell lines exist in at least two maturation states, characterized by the onset of insulin 1 promoter activity.1, 3 Our recently published microarray analysis of Pdx1+/Inslow immature and Pdx1+/Ins+ mature human β-cells pointed to an enrichment of Musashi in immature β-cells.3 We have also shown that activin A can dedifferentiate adult β-cells in culture.2 Here, we demonstrate that treatment of primary mouse islets and MIN6 β-cells with activin A significantly increased the expression of Msi1 (Figures 4a and b). Thus, Musashi genes are present and can be dynamically regulated in conjunction with the differentiation status of adult islet cells.

Musashi expression increases with decreased β-cell differentiation status. (a) RT-qPCR analysis of Msi1 and Msi2 expression levels in mouse islets (n=4) and (b) MIN6 cells after treatment with 4 nM of activin A for 48 h (n=6). *P<0.05
ER stress and lipotoxicity regulate Musashi and Notch pathway gene expression
Type 2 diabetes is characterized by pancreatic β-cell dedifferentiation and dysfunction.23, 24, 25 Interestingly, our gene expression analysis showed that HES1 expression was significantly increased in type 2 diabetic islets compared to non-diabetic controls (Figure 5a). Obesity and free fatty acids increase the risk of type 2 diabetes, in part by inducing β-cell dedifferentiation, dysfunction and apoptosis.26, 27 Thus, we investigated whether hyperglycemia or hyperlipidemia might alter the expression of Musashi genes. Musashi gene expression was unaffected by varying the glucose concentration (Figure 5b). In contrast, lipotoxic conditions induced by the free fatty acid palmitate significantly increased Msi2 (Figure 5c). Consistent with the increase in Musashi, palmitate induced Hes1 1.8-fold (Figure 5c). Many of the deleterious effects of palmitate on β-cell function and survival are mediated by ER stress. Blocking ER Ca2+ influx with thapsigargin28 was sufficient to induce a significant upregulation of Msi1, Msi2 and Hes1 mRNA levels (Figures 5d–f and k). Msi2 protein levels were also significantly increased with thapsigargin treatment (Figures 5g and h). As expected, both insulin genes were highly downregulated with thapsigargin-induced ER stress (Figures 5d, m and n). Thapsigargin had opposing effects on Notch1 and Notch2 (Figures 5i and j). Surprisingly, the γ-secretase inhibitor DAPT did not prevent these effects of thapsigargin, suggesting that the regulation of Ins1, Ins2, Hes1 and Ngn3 by ER stress is independent of classical Notch signaling under these experimental conditions (Figures 5e, f and i–n). In keeping with a Notch-independent mechanism, we did not see changes in the protein levels of Numb (Figure 5o), a Notch inhibitor and putative target of Musashi translational regulation. Taken together, these data demonstrate that lipotoxicity and ER stress upregulate Musashi and Hes1 via a non-canonical pathway and suggest a modulatory role for this pathway in type 2 diabetes. Interestingly, we did not observe changes in Msi1 or Msi2 expression with a cocktail of pro-inflammatory cytokines designed to mimic conditions in type 1 diabetes (Figures 5p and q), indicating that the role of Musashi is relatively specific to dedifferentiation associated with lipotoxic/ER stress conditions in type 2 diabetes.

Lipotoxicity and ER stress induce changes in Musashi and the Notch pathway. (a) RT-qPCR analysis of MSI1, MSI2 and HES1 gene expression levels in non-diabetic (n=8) and type 2 diabetic human islets (n=3). (b) MIN6 cells were treated with increasing glucose concentrations for 24 h, followed by RT-qPCR analysis of Msi1 and Msi2 mRNA levels (n=3). (c) MIN6 cells were treated with 1.5 mM of the free fatty acid palmitate for 5, 9 or 24 h to induce a lipotoxic stress after which Msi1, Msi2 and Hes1 mRNA levels were quantified by RT-qPCR. Expression is normalized to untreated control samples (n=4). (d) MIN6 cells were treated with 1 μM thapsigargin for 5, 9 or 24 h, followed by RT-qPCR for Msi1, Msi2, Ins1, Ins2 and Hes1. (e–q) MIN6 cells were treated with 5 μM DAPT, 1 μM thapsigargin or both for 24 h (n=4), followed by RT-qPCR analysis of Msi1, Msi2 Hes1, Ngn3, Notch1, Notch2, Ins1 and Ins2 mRNA levels (e, f and i–n) and immunoblot for Msi1, Msi2 and Numb (g, h and o). (p and q) MIN6 cells were treated with a cytokine cocktail containing TNF-α (25 ng/ml), IL-1β (10 ng/ml) and IFN-γ (10 ng/ml) for 8 h (n=4). (r and s) Overexpression of MSI1:eGFP protects MIN6 cells from cytokine- and thapsigargin-induced apoptotic cell death. Propidium iodide (PI) incorporation was used as an index of cell death (r), while AnnexinV positivity was used as an index of caspase-3-dependent apoptosis. Controls are overexpressing an eGFP vector. * P<0.05 versus untreated #P<0.05 versus eGFP controls
Musashi 1 regulates β-cell proliferation and insulin gene transcription
Thus far, our studies showed that Musashi levels were associated with changes in β-cell differentiation status, but they did not inform us as to whether either of the Musashi isoforms could drive some or all aspects of this process. To address the function of Musashi, we used overexpression and knockdown in MIN6 cells. Transfection of human MSI1:GFP resulted in significant Musashi-1 overexpression, with no change in Msi2 (Figure 6a). Given the induction of Msi1 under stress conditions and its known antiapoptotic roles in other cell types,29 we first assessed the effect of MSI:GFP overexpression on basal and induced β-cell apoptosis. Indeed, overexpression of MSI1 strongly reduced basal apoptosis and programmed cell death induced by proinflammatory cytokines and thapsigargin (Figures 5r and s). Remarkably, overexpression of MSI1 alone was sufficient to significantly decrease Ins2 gene expression (Figure 6b). On the other hand, shRNA-mediated Msi1 knockdown had only a slight effect on Ins2 (Figures 6f and g), suggesting that this Musashi isoform is not a necessary regulator of basal insulin transcription. Taken together, these data demonstrated that Musashi can drive changes in β-cell insulin gene expression. We have previously shown that immature and dedifferentiated states are associated with increased β-cell proliferation.1, 2 MSI1 overexpression significantly increased MIN6 cell proliferation (Figures 6c and d), while Msi1 knockdown had a negative effect on proliferation (Figures 6h and i). This result is consistent with the role of Msi1 in promoting proliferation of intestinal progenitors.30 As expected, MSI1 overexpression significantly increased Hes1 expression (Figure 6e) and Msi1 knockdown significantly decreased Hes1 gene expression (Figure 6j), suggesting that Musashi regulates Hes1 in adult β-cells, similar to other systems.

Msi1 regulates insulin gene expression and β-cell proliferation via Hes1. MIN6 cells overexpressing human MSI1-GFP and GFP control plasmid were analyzed for (a) Msi1 and Msi2 (n=4) (b) and Ins1 and Ins2 (n=4) mRNA levels by RT-qPCR. (c) Representative images of MIN6 cells transfected with a GFP control plasmid (top) or MSI1-GFP plasmid (bottom), stained for BrdU and quantified (d) (n=3). (e) Hesl mRNA levels in MSI1-overexpressing MIN6 cells (n=4). RT-qPCR analysis of (f) Msi1 and Msi2 (n=7) and (g) Ins1 and Ins2 (n=6) mRNA levels in MIN6 cells transfected with RFP-tagged Msi1-targeted shRNA versus RFP scramble control. (h) Representative images of BrdU-stained MIN6 cells transfected with RFP scramble control plasmid (top) or RFP-tagged shRNA-targeting Msi1 (bottom) and quantified (i) (n=4). (j) Hes1 gene expression in MIN6 cells with Msi1 knockdown (n=7). *P<0.05
Although Msi1 and Msi2 are homologs, the differences in their expression levels suggested the possibility of non-overlapping functions. MSI2 overexpression significantly decreased Ins1 and Ins2 gene expression (Figures 7a and b), whereas Msi2 knockdown significantly increased Ins2 expression (Figures 7f and g), similar to the findings with Msi1. However, in contrast to the effects of MSI1, MSI2 overexpression blocked MIN6 proliferation (Figures 7c and d), whereas Msi2 knockdown augmented MIN6 proliferation (Figures 7h and i). This was associated with and perhaps caused by a compensatory effect on Msi1 expression with MSI2 overexpression or knockdown (Figures 7a and f). Unexpectedly, both MSI2 overexpression and Msi2 knockdown increased Hes1 gene expression (Figures 7e and j). This suggests that Hes1 expression is uncoupled from the proliferative effects of Musashi and may be regulated exclusively by Msi1. Taken together, these data suggest that the Msi1 and Msi2 play overlapping, but distinct, roles in the control of β-cell proliferation and insulin gene expression and may also regulate the expression of each other under some conditions.

Msi2 regulates insulin gene expression and has reciprocal effects on proliferation and Hes1 gene expression compared to Msi1. MIN6 cells overexpressing human MSI2-mCherry and mCherry control plasmid were analyzed for (a) Msi1 and Msi2 (n=4) and (b) Ins1 and Ins2 (n=3) mRNA levels by RT-qPCR. (c) Representative images of MIN6 cells transfected with mCherry control plasmid (top) or MSI2-mCherry plasmid (bottom), stained for BrdU and quantified (n=4) (d). (e) Hesl mRNA levels in MSI2-overexpressing MIN6 cells (n=3). MIN6 cells transfected with GFP-tagged-shRNA-targeting Msi2 or GFP scramble control were analyzed for (f) Msi1 and Msi2 gene expression (n=3) and (g) Ins1 and Ins2 (n=3) mRNA levels by RT-qPCR. (h) Representative images of MIN6 cells transfected with GFP scramble control shRNA (top) or GFP-tagged Msi2-targeted shRNA (bottom) and stained for BrdU and quantified (n=3) (i). (j) Hes1 mRNA levels in MIN6 cells with Msi2 knockdown (n=3). *P<0.05
Discussion
The purpose of this study was to characterize the presence and function of the Musashi genes in pancreatic β-cells. We showed that both isoforms of Musashi were abundant in adult pancreatic α- and β-cells. Both proteins were also present in human fetal pancreas and during the directed differentiation of hESCs towards the pancreatic lineage, but the expression pattern was not consistent with that of a stem cell marker. Musashi expression was also elevated with a decreased differentiation status in adult β-cells and conditions of ER stress. Musashi overexpression and knockdown experiments pointed to a suppressive role in insulin gene expression. Msi1, but not Msi2, exhibited a proliferative effect. We identify a role for the known Notch target Hes1, but surprisingly find that its induction is independent of Numb protein levels or Notch activation (Figure 8).

Working model of Musashi regulation and action in adult β-cells. Our data suggest that Musashi-1 and Musashi-2 are differentially regulated by upstream signals, and possibly each other that Musashi-1 and Musashi-2 regulate insulin expression, proliferation and apoptosis, via Notch- and Numb-independent signaling. This is associated with changes in Hes1 mRNA, suggesting that this downstream gene may play a role in the mechanism
The Notch/Hes1/Ngn3 pathway plays important roles in the development of pancreatic endocrine cells. During pancreatic development, temporally regulated Ngn3 expression is required for the differentiation of all endocrine lineages.6, 31 Ngn3 is regulated by Notch signaling through repression by the Notch target gene Hes1.7 Hence, the Notch/Ngn3 signaling pathway sustains the proliferating pool of pancreatic progenitor cells and their eventual exocrine or endocrine cell fates.8 It has become increasingly evident that the developmentally critical Notch/Ngn3 signaling pathway is also active in adult islets, but the upstream regulators of this pathway in adult islets remain unclear. Ngn3 has been shown to be expressed, albeit at very low levels, in adult islets at the mRNA and protein levels.9, 10 We have previously also shown that inhibiting Notch signaling in adult β-cells increased Ngn3 expression and β-cell apoptosis in a context-dependent manner, while NGN3 overexpression was sufficient to induce β-cell apoptosis.9 This fits with our current data showing antiapoptotic roles for Musashi, a negative regulator of Ngn3. Others later showed using conditional gene knockout that Ngn3 plays a role in adult β-cell maturation and function.10 In this study, we have further confirmed that several components of the Notch signaling pathway are expressed and active in adult β-cells, although the signaling between Musashi and Hes1 does not appear to follow the classical Notch pathway. Regardless of this atypical mechanism, our data suggest that Musashi, and perhaps Hes1, contribute to the modulation of β-cell proliferation and maturation. Our overexpression and knockdown data also demonstrate that each of the Musashi genes have generally reciprocal effects on proliferation and Hes1 gene expression, suggesting that they may have some distinct functional roles. Our overexpression data also suggest that Msi2 negatively regulates Msi1.14 Collectively, our studies point to a stronger role for Msi1 in regulating proliferation, modulating β-cell survival and activating Hes1 expression, whereas both Musashi genes seem to regulate insulin gene expression in the same manner.
We have previously described an immature subpopulation of adult β-cells in humans and mice marked by Pdx1 promoter activity with little or no insulin-1 promoter activity.1, 2, 3 MSI2 was significantly increased in purified immature human β-cells.3 Here, a subset of adult islets and β-cells strongly expressed the Musashi genes. The dedifferentiation and hyperproliferation of adult β-cells by activin A was associated with an increase in Msi1. Furthermore, our results support the notion that islets from type 2 diabetes patients have a dedifferentiated phenotype23 with increased HES1. Our knockdown experiments suggest that Musashi acts as a constitutive break on insulin expression in mature β-cells, similar to what we have previously shown with follistatin.2 The idea that Musashi may be associated with an immature β-cell phenotype in adult islets is also consistent with its role in other systems, including a putative breast stem cell population.12, 32 In the central nervous system, both Musashi isoforms were described to be highly enriched and play a role in the maintenance of neural stem cells, becoming downregulated with differentiation.14 The intestinal crypt, which contains the highly proliferative stem cell pool, was also characterized by the expression of both Msi1 and Hes1.15 This is consistent with our findings that MSI1 overexpression increased the expression of Hes1 as well as proliferation.
Numerous studies have reported multi-hormonal cells in the embryonic and fetal pancreas,33, 34, 35 but the factors that govern this state are unclear. In human fetal pancreas, we found that both Musashi isoforms were present in cells that expressed insulin and glucagon or both. While early studies employing toxin-mediated lineage reduction suggested that the majority of adult mouse β-cells had not previously expressed the glucagon promoter,36 more recent data point to the production of β-cells under extreme stress conditions from cells with transient glucagon promoter activity.5 Our group has recently characterized a large population of bihormonal cells in the human fetal pancreas and provided histological evidence that they may give rise to mature α-cells.22 This observation mirrored the multi-hormonal cells seen after directed hESC differentiation.21 Single-cell transcript analysis supports existence of islet cells co-expressing multiple hormones, including insulin and glucagon.37, 38 Microarray analysis of immature versus mature β-cells from humans, mice and MIN6 cells revealed that the immature cells were multi-hormonal.3 Considering that MSI2 was also significantly upregulated in immature human β-cells, our results support the concept that elevated Musashi may be found in an immature cell state in the adult pancreas and perhaps the possibility of expressing multiple hormones.
We have identified Musashi as a possible link between ER stress to the downregulation of insulin expression seen in the type 2 diabetes and lipotoxicity.39 Given that Musashi appears to represent a compensatory protective gene, it is possible that it could transduce the effects of ER stress and lipotoxicity into an increase in β-cell proliferation and β-cell mass typically seen in the early stages of type 2 diabetes.23, 24, 25 It is well established that ER stress is associated with general translational inhibition, but our study now implicates Musashi, a translational repressor, in the stress-dependent control of the insulin gene. Analysis of publically accessible resources and our own expression studies demonstrated that the Musashi/Hes1 pathway may be increased in islets from patients with type 2 diabetes. Although multiple stresses affect islets in the complex milieu of diabetes, we demonstrated in vitro that lipotoxicity with the free fatty acid palmitate is sufficient to recapitulate the increase in Musashi and Hes1 gene expression. Further, we demonstrated that the ER stress component of palmitate-induced lipotoxicity is sufficient for the increase in Musashi levels. However, our data suggest that ER-stress-induced effects on Hes1 may be Numb- and Notch-independent. Hence, other Musashi targets likely link Musashi and Hes1. Taken together with our observations that increases in Musashi were sufficient to reduce insulin expression, inhibit apoptosis and increase β-cell proliferation, these data suggest that Musashi may be involved in the decreased level of insulin expression and possibly the subsequent β-cell dedifferentiation seen in type 2 diabetes.
Collectively, our results are the first to demonstrate that Musashi is present in developing and adult β-cells, where it is responsive to multiple external factors that regulate cellular differentiation status, including ER stress. Our data point to differences in the functional roles of Musashi-1 and Musashi-2, while suggesting a conserved role for this gene family as an upstream regulator of Hes1. Our gain-of-function and loss-of-function studies demonstrate that Musashi is necessary and sufficient to suppress β-cell proliferation and insulin gene expression. Musashi-1 and Musashi-2 are novel regulators of β-cell function and fate.
Materials and Methods
Reagents
Respective RFP- and GFP-bearing shRNA constructs targeted to Msi1 and Msi2 were purchased from OriGene (Rockville, MD, USA). Overexpression constructs for human MSI1-GFP and GFP control plasmids were purchased from OriGene. Human MSI2-mCherry and mCherry control plasmids were purchased from GeneCopoeia (Rockville, MD, USA). These were introduced to cells by transfection (Lipofectamine 2000; Invitrogen, Burlington, ON, USA) or in later experiments by the Invitrogen Neon instrument. Recombinant human activin A was purchased from R&D Systems (Minneapolis, MN, USA). Thapsigargin was from Sigma (Oakville, ON, USA). Palmitate (Sigma) was made up as a complex to BSA as described previously.26 The γ-secretase inhibitor of Notch activation, DAPT, was purchased from Sigma.
Cell culture
All cell culture reagents were from Invitrogen, unless otherwise stated. Human islets were provided by the Ike Barber Human Islet Transplant Laboratory (Vancouver General Hospital, Vancouver, BC, Canada). Mouse islets were isolated from 10–12-week-old C57BL/6J mice using collagenase and filtration. Islets were hand-picked and cultured overnight at 37 °C and 5% CO2 in RPMI1640 medium supplemented with 5 mM glucose (for human islets) and 11 mM glucose (for mouse islets), 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine serum (Invitrogen). All animal protocols were approved by the University of British Columbia, in accordance with national guidelines. MIN6 cells were cultured as described.1
Pancreatic differentiation of hESCs
HESCs derived from the H1 line and a variant of the CA1 line, CA1S, were maintained as undifferentiated populations on 1 : 30 diluted Matrigel (BD Biosciences, Bedford, MA, USA) in mTesR1 maintenance medium (Stem Cell Technologies, Vancouver, BC, Canada) as described elsewhere.40 Cells were passaged every 3–7 days by passaging 1 : 3 to 1 : 6 to maintain culture densities between 30 and 80% confluent. Pancreatic differentiation of hES cells was carried out following the stage-specific differentiation methods described previously.21
Gene expression analysis
Bioinformatic analysis of gene expression was carried out using our previously described approach.19 For RT-PCR and qRT-PCR, total RNA was purified using the RNeasy kit (Qiagen, Mississauga, ON, Canada). cDNA was prepared from 200 to 600 ng of RNA using the qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD, USA). Primers were designed to flank an intron and are available on request. qRT-PCR was performed by using the PerfeCTa SYBR Green SuperMix with ROX (Quanta Biosciences) and Step One Plus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). For relative quantification of transcripts, Ct values for each sample were normalized to GAPDH and ALDOA for human samples, and HPRT1 and β-actin for mouse samples, using the Data Assist software (Applied Biosystems). RT-PCR was performed using Taq polymerase (Fermentas, Burlington, ON, Canada).
Immunoblot
MIN6 cells, mouse islets, mouse exocrine tissue and mouse brain tissue were lysed in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). All other immunoblot methods were performed as described,19 using primary antibodies as follows: mouse anti-β-actin (1 : 1000; Novus Biologicals, Littleton, CO, USA), rabbit anti-Musashi-1 and -2 (1 : 500; Millipore, Billerica, MA, USA), mouse anti-Numb/Cnmb-1 (1 : 200; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA) and rabbit anti-Notch1 (1 : 1000; Cell Signaling Technology).
Online analysis of single β-cell apoptosis
Cell death and apoptosis were tracked via the nuclear incorporation of propidium iodide (PI; near red; all forms of cell death) and AlexaFluor647-conjugated AnnexinV (far red; apoptosis) binding to the outer membrane (and eventually entering the cell). Nuclear morphology and cell numbers were visualized with a low concentration of Hoechst. The number of control (eGFP only) and MSI:GFP-overexpressing cells that also expressed PI and/or AnnexinV were counted automatically using the Molecular Devices ImageXpressMICRO high-content screening instruments.
Fluorescence staining
Human islets and pancreata from adult donors between 23 and 56 years old of either sex were obtained from consenting donors via the Clinical Islet Transplantation Program at the Vancouver General Hospital and were not from diabetic donors, unless otherwise indicated. Human fetal pancreas sections were kindly provided by Dr. Rennian Wang (University of Western Ontario, London, ON, Canada). Pancreas sections were 5 μm. Isolated mouse pancreata and human fetal tissues were fixed in 4% paraformaldehyde (PFA) overnight at 4 °C, rinsed in 70% ethanol and embedded in paraffin. The tissue was sectioned for immunostaining at 5 μm thickness. Staining of mouse and human adult and fetal pancreas sections was performed as described previously.21 MIN6 cells were seeded at low density on coverslips for single-cell staining before fixing in PFA. Polyclonal rabbit-anti-Musashi-1 (1 : 200) and rabbit-anti-Musashi-2 (1 : 200) were purchased from Millipore. Polyclonal guinea-pig anti-insulin (1 : 1000) was from Millipore. Mouse anti-glucagon (1 : 1000) was from Sigma (St. Louis, MO, USA). Images were taken on the ImageXpressMICRO using high NA objectives and analyzed using the MetaXpress software (Molecular Devices Corporation, Sunnyvale, CA, USA).
5-Bromo-2-deoxyuridine (BrdU) labeling was performed on MIN6 cells overexpressing Musashi fusion proteins or fluorescent protein-bearing shRNA constructs purified by FACS. BrdU (10 μM; Kit I; Roche, Laval, QC, Canada) was added to FACS-purified MIN6 cells for 3–6 h before fixation. Cells were imaged using a Zeiss 200 M microscope equipped with a FLUAR × 10 and × 100 (1.45 NA) objectives (Carl Zeiss, Thornwood, NY, USA) and ImageXpressMICRO. Images were analyzed and deconvolved using SlideBook (Intelligent Imaging Innovations, Boulder, CO, USA) or AcuityXpress (Molecular Devices Corporation). A minimum of 500 cells per sample were quantified.
Data analysis
Data are presented as means±S.E.M. Differences between means were evaluated by Student's unpaired t-tests or one-way ANOVA, as appropriate. A P-value of 0.05 or less was considered significant. A minimum of three independent experiments were performed for each study.
Abbreviations
- hESCs:
-
human embryonic stem cells
- ER:
-
endoplasmic reticulum
- DAPT:
-
N-[(3,5-difluorophenyl)acetyl]-L-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester
- RT-qPCR:
-
reverse transcriptase-quantitative polymerase chain reaction
- MIN6:
-
mouse insulinoma cell line 6
- BrdU:
-
5-bromo-2-deoxyuridine
References
Szabat M, Luciani DS, Piret JM, Johnson JD . Maturation of adult beta-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology 2009; 150: 1627–1635.
Szabat M, Johnson JD, Piret JM . Reciprocal modulation of adult beta cell maturity by activin A and follistatin. Diabetologia 2010; 53: 1680–1689.
Szabat M, Pourghaderi P, Soukhatcheva G, Verchere CB, Warnock GL, Piret JM et al. Kinetics and genomic profiling of adult human and mouse β-cell maturation. Islets 2011; 3: 175–187.
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA . In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008; 455: 627–632.
Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010; 464: 1149–1154.
Gradwohl G, Dierich A, LeMeur M, Guillemot F . Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 2000; 97: 1607–1611.
Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang JH et al. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes 2001; 50: 928–936.
Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T et al. Notch signalling controls pancreatic cell differentiation. Nature 1999; 400: 877–881.
Dror V, Nguyen V, Walia P, Kalynyak TB, Hill JA, Johnson JD . Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia 2007; 50: 2504–2515.
Wang S, Jensen JN, Seymour PA, Hsu W, Dor Y, Sander M et al. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc Natl Acad Sci USA 2009; 106: 9715–9720.
Imai T, Tokunaga A, Yoshida T, Hashimoto M, Mikoshiba K, Weinmaster G et al. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol Cell Biol 2001; 21: 3888–3900.
Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS . A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol 2005; 277: 443–456.
McGill MA, McGlade CJ . Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem 2003; 278: 23196–23203.
Sakakibara S, Nakamura Y, Yoshida T, Shibata S, Koike M, Takano H et al. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci USA 2002; 99: 15194–15199.
Kayahara T, Sawada M, Takaishi S, Fukui H, Seno H, Fukuzawa H et al. Candidate markers for stem and early progenitor cells, Musashi-1 and Hes1, are expressed in crypt base columnar cells of mouse small intestine. FEBS Lett 2003; 535: 131–135.
Berdnik D, Torok T, Gonzalez-Gaitan M, Knoblich JA . The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev Cell 2002; 3: 221–231.
Yoshida T, Tokunaga A, Nakao K, Okano H . Distinct expression patterns of splicing isoforms of mNumb in the endocrine lineage of developing pancreas. Differentiation 2003; 71: 486–495.
Hoffman BG, Robertson G, Zavaglia B, Beach M, Cullum R, Lee S et al. Locus co-occupancy, nucleosome positioning, and H3K4me1 regulate the functionality of FOXA2-, HNF4A-, and PDX1-bound loci in islets and liver. Genome Res 2010; 20: 1037–1051.
Yang YH, Szabat M, Bragagnini C, Kott K, Helgason CD, Hoffman BG et al. Paracrine signalling loops in adult human and mouse pancreatic islets: netrins modulate beta cell apoptosis signalling via dependence receptors. Diabetologia 2011; 54: 828–842.
Okano H, Imai T, Okabe M . Musashi: a translational regulator of cell fate. J Cell Sci 2002; 115: 1355–1359.
Rezania A, Riedel MJ, Wideman RD, Karanu F, Ao Z, Warnock GL et al. Production of functional glucagon-secreting alpha-cells from human embryonic stem cells. Diabetes 2011; 60: 239–247.
Riedel MJ, Asadi A, Wang R, Ao Z, Warnock GL, Kieffer TJ . Immunohistochemical characterization of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia 2011; e-pub ahead of print 25 0ctober 2011.
Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005; 54: 727–735.
Ahren J, Ahren B, Wierup N . Increased beta-cell volume in mice fed a high-fat diet: a dynamic study over 12 months. Islets 2010; 2: 12–15.
Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng YH et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 2005; 122: 337–349.
Jeffrey KD, Alejandro EU, Luciani DS, Kalynyak TB, Hu X, Li H et al. Carboxypeptidase E mediates palmitate-induced beta-cell ER stress and apoptosis. Proc Natl Acad Sci USA 2008; 105: 8452–8457.
Unger RH . Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995; 44: 863–870.
Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 2009; 58: 422–432.
Sureban SM, May R, George RJ, Dieckgraefe BK, McLeod HL, Ramalingam S et al. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology 2008; 134: 1448–1458.
Rezza A, Skah S, Roche C, Nadjar J, Samarut J, Plateroti M . The overexpression of the putative gut stem cell marker Musashi-1 induces tumorigenesis through Wnt and Notch activation. J Cell Sci 2010; 123: 3256–3265.
Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell 2007; 12: 457–465.
Wang XY, Yin Y, Yuan H, Sakamaki T, Okano H, Glazer RI . Musashi1 modulates mammary progenitor cell expansion through proliferin-mediated activation of the Wnt and Notch pathways. Mol Cell Biol 2008; 28: 3589–3599.
Lukinius A, Ericsson JL, Grimelius L, Korsgren O . Ultrastructural studies of the ontogeny of fetal human and porcine endocrine pancreas, with special reference to colocalization of the four major islet hormones. Dev Biol 1992; 153: 376–385.
Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D . Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development 1993; 118: 1031–1039.
Herrera PL, Huarte J, Sanvito F, Meda P, Orci L, Vassalli JD . Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development 1991; 113: 1257–1265.
Herrera PL, Huarte J, Zufferey R, Nichols A, Mermillod B, Philippe J et al. Ablation of islet endocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc Natl Acad Sci USA 1994; 91: 12999–13003.
Katsuta H, Akashi T, Katsuta R, Nagaya M, Kim D, Arinobu Y et al. Single pancreatic beta cells co-express multiple islet hormone genes in mice. Diabetologia 2009; 53: 128–138.
Chiang MK, Melton DA . Single-cell transcript analysis of pancreas development. Dev Cell 2003; 4: 383–393.
Fontes G, Semache M, Hagman DK, Tremblay C, Shah R, Rhodes CJ et al. Involvement of Per-Arnt-Sim kinase and extracellular-regulated kinases-1/2 in palmitate inhibition of insulin gene expression in pancreatic beta-cells. Diabetes 2009; 58: 2048–2058.
Schulze HG, Konorov SO, Caron NJ, Piret JM, Blades MW, Turner RF . Assessing differentiation status of human embryonic stem cells noninvasively using Raman microspectroscopy. Anal Chem 2010; 82: 5020–5027.
Acknowledgements
We thank Dr. Rennian Wang (University of Western Ontario, Canada) for providing human fetal tissue samples. We also thank Dr. Dan Luciani for providing insight into this manuscript. We also thank Mr. Kevin Tsai for technical assistance on qPCR experiments. This work was supported by an operating grant to JDJ and TJK from the Stem Cell Network and from the Canadian Institutes for Health Research (Regeneration and Nanomedicine Initiative). JDJ was supported by salary awards from the Juvenile Diabetes Research Foundation, Canadian Diabetes Association, Michael Smith Foundation for Health Research and the Canadian Institutes for Health Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Szabat, M., Kalynyak, T., Lim, G. et al. Musashi expression in β-cells coordinates insulin expression, apoptosis and proliferation in response to endoplasmic reticulum stress in diabetes. Cell Death Dis 2, e232 (2011). https://doi.org/10.1038/cddis.2011.119
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.119
Keywords
This article is cited by
-
Roles and regulation of tRNA-derived small RNAs in animals
Nature Reviews Molecular Cell Biology (2024)
-
MSI1 Stabilizes MACF1 to Inhibit Apoptosis of MC3T3-E1 Cells Induced by High Glucose and Promote Osteogenic Differentiation Through Wnt/β-Catenin Signaling Pathway
Molecular Biotechnology (2023)
-
Linking Diabetes Mellitus with Alzheimer's Disease: Bioinformatics Analysis for the Potential Pathways and Characteristic Genes
Biochemical Genetics (2022)
-
Musashi 2 influences chronic lymphocytic leukemia cell survival and growth making it a potential therapeutic target
Leukemia (2021)
-
A new shortened protocol to obtain islet-like cells from hESC-derived ductal cells
In Vitro Cellular & Developmental Biology - Animal (2021)
