
Abstract
Although much is known about transcriptional networks that control embryonic stem cell (ESC) self-renewal and differentiation, the metabolic regulation of ESC is less clear. Autophagy is a catabolic process that is activated under both stress and normal conditions to degrade damaged organelles and aggregated proteins, and thus plays pivotal roles in somatic and adult stem cell function. However, if and how ESCs harness autophagy to regulate stemness remains largely unknown. Recently, we have defined that autophagy is essential for mitochondrial homeostasis regulation in pluripotency acquirement and maintenance. Here we identified high autophagic flux as an essential mechanism to maintain ESC identity. We show that mouse ESCs exhibit a high autophagic flux that is maintained by coordinating expression of autophagy core molecular machinery genes through FOXO1, a forkhead family transcription factor. Tapering autophagic flux by manipulating either Atg3 or Foxo1 expression compromised ESC self-renewal, pluripotency, and differentiation that could be restored by gain of wild-type but not function-deficient Atg3 or Foxo1 mutants, respectively. Our results define a newly recognized role of autophagic flux in mouse ESC identity maintenance that links cellular catabolism to ESC fate regulation.
Similar content being viewed by others


Main
Whereas much is known about the transcriptional networks controlling embryonic stem cell (ESC) self-renewal and differentiation activities, less is understood on metabolic regulation of ESCs. Macroautophagy (hereafter referred to autophagy) is a catabolic process wherein both organelles and proteins are degraded to maintain cellular homeostasis.1, 2, 3 The autophagy process is regulated by mTOR (mammalian target of rapamycin) and a set of autophagy-related genes. The mTOR, a serine/threonine protein kinase, negatively regulates autophagy by inhibitory phosphorylation of ULK1 (S757) and preventing formation of ULK1–ATG13–FIP200 complex (which is required for autophagy initiation) through mTOR complex 1 (mTORC1).4, 5, 6, 7 Basal autophagy is required to metabolize cytoplasmic components and avoid accumulation of damaged proteins and organelles and plays important roles in mammalian differentiation and development.8
Autophagy has been shown to play important roles in adult stem cells. For example, competent autophagy was demonstrated to protect hematopoietic stem cell from metabolic stress by a FOXO3A-driven pro-autophagy gene program.9 In muscle stem cells, autophagy maintains stemness by preventing senescence.10 Pluripotent stem cells (PSCs), including induced pluripotent and ESCs, hold great promise for regenerative medicine because of their ability to undergo unlimited self-renewal and retain pluripotency to differentiate into all cell types of the three germ layers.11, 12, 13, 14 Recent studies have shown that both canonical and noncanonical autophagy plays important roles for reprogramming to pluripotency, indicating the potential involvement of autophagy for stemness regulation in ESCs.15, 16, 17 Most recently, we have demonstrated that canonical autophagy regulates mitochondrial homeostasis in pluripotency acquirement and maintenance. At the same time we have provided evidences to show that canonical autophagy involves in mouse ESC self-renewal and pluripotency.18 However, the characters of autophagy in ESC and how does autophagy regulate ESC stemness is largely unknown.
Results
PSCs have high autophagic flux
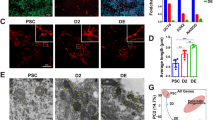
To investigate how autophagy regulates ESC identity, we first compared autophagic flux between somatic cells (mouse embryonic fibroblasts (MEFs); ESC-derived fibroblasts; ESC-derived neuron stem cells), ESCs, and MEF-derived iPSCs. Autophagic flux was determined by assaying LC3-II turnover in cells that were either untreated or treated with the autophagy inhibitor chloroquine (Supplementary Figure S1c). Although MEFs have a higher LC3-II/LC3-I ratio than that of PSCs, the LC3-II enrichment extent in chloroquine-treated PSCs is significantly higher than that in somatic fibroblasts, indicating that PSCs have a higher basal autophagic flux (Figures 1a, b, and e and Supplementary Figures S1a and b). Next, we tracked changes in autophagosome number and morphology using transmission electronic microscopy (TEM). Although the number of autophagosomes in PSCs was lower than that in MEFs (Figure 1c and Supplementary Figure S1d), the increase of autophagosomes in PSCs upon chloroquine treatment is significantly higher than that of MEFs (Figure 1d). Furthermore, the percentage of autophagy vacuole (AV) areas in PSCs upon chloroquine treatment is significantly higher than that of MEFs (Supplementary Figures S1e and f). These results are consistent with western blot data (Figure 1a). We then investigated differences in autophagy induction in cells following starvation. The accumulation of LC3-II in PSCs was significantly higher than that of MEFs upon starvation treatment, indicating that PSCs have a powerful autophagic infrastructure that can provide enhanced autophagic potency upon induction (Figure 1f and Supplementary Figures S2a and b).

High autophagic flux in pluripotent stem cells. (a) Western blotting for LC3 and β-actin in B6/MEF, B6/ES, and 1E/iPS cells. β-Actin served as the loading control. CQ, chloroquine. CQ treatment: 50 μM, 5 h. (b) Quantification of autophagic flux by calculating the ratio of LC3-II with or without CQ treatment in (a). Data shown are the mean±S.D., n=4, *P<0.05, **P<0.01 (Student’s t-test). (c) Representative TEM images of autophagic vacuoles (AVs, indicated by white arrowheads) in B6/MEF, B6/ES, and 1E/iPS cells with or without CQ treatment (50 μM, 5 h); scale bars, 2 μm. (d) Quantification of autophagic flux calculated from the ratio of the number of AVs in untreated and CQ treated cells in (c). Mean±S.D. from three independent experiments are shown and ~20 cells were counted. **P<0.01, ***P<0.001 (Student’s t-test). (e) Western blotting for LC3 and β-actin in ESC, ESC-derived NSC, and ESC-derived fibroblast. β-Actin served as the loading control. CQ, chloroquine. CQ treatment: 50 μM, 5 h. (f) Western blotting for LC3 and actin in B6/MEF, B6/ES, and 1E/iPS cells under basal or starvation conditions. S, starvation. Cells underwent EBSS starvation for 5 h (these conditions were used for similar experiments). CQ treatment: 100 μM, 5 h. (g) Time-course analysis of degradation of long-lived proteins in B6/MEF, B6/ES, and 1E/iPS cells under starvation conditions (EBSS starvation for 2 or 5 h). Inhibition of degradation of long-lived proteins by treatment with 10 mM 3-methyladenine (3-MA) for 2 or 5 h. Data shown are mean±S.D., n=3, *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). Images in (a, e, and f) are representative of three independent experiments
Long-lived protein degradation is currently the most quantitative assay to determine autophagic flux, because it provides a precise numerical readout that reflects the fate of all long-lived cellular proteins but not a single autophagic substrate.19 To further confirm the differential autophagic fluxes between PSCs and somatic cells, we measured the bulk degradation of long-lived proteins. Cellular proteins were labeled with 14C for 24 h, followed by 1 h of incubation without 14C-labeled amino acids to exclude short-lived radiolabeled proteins that are usually degraded by the proteasome.20, 21 The cellular release of degraded proteins under the different treatment conditions was then quantified. In agreement with the high autophagic flux observed in PSCs, these cells have a significantly higher protein degradation rate relative to MEFs (Figure 1g). With starvation treatment, the PSCs induced robust degradation of long-lived proteins with much faster dynamics than that of MEFs, in which degradation could be inhibited by 3-methyladenine (3-MA)/bafilomycin A1 (Baf-A1) treatment (Figure 1g and Supplementary Figure S2c). Collectively, these data demonstrate that, compared with somatic fibroblasts, PSCs not only have a higher basal autophagic flux but also a higher potency for autophagy induction in response to stress.
High autophagic flux in PSCs is maintained by enhanced autophagy core machinery gene expression
To investigate the mechanisms by which PSCs maintain high autophagic flux and robust competency for autophagy induction, we next tested the activation status of mTORC1, the principal pathway that inhibits autophagy induction.2, 22, 23 We saw no differences in mTORC1 activation between PSCs and MEFs, as detected by measuring levels of phosphorylated mTOR (p-mTOR) or phosphorylated p70S6K (p-p70S6K), the two direct targets of mTORC1 in autophagy regulation (Figure 2a). These data indicate that the increased autophagic flux in PSCs is not due to differential mTORC1 activation between PSCs and somatic fibroblasts.

Enhanced expression of autophagy core molecular machinery genes in PSCs. (a) Western blotting for phosphorylated mTOR (p-mTOR), mTOR, phosphorylated p70S6K (p-p70S6K), and p70S6K in B6/MEF, B6/ES, and 1E/iPS cells. β-Actin served as the loading control. (b– f) qPCR for autophagy core molecular machinery genes sorted into five complexes in B6/MEF, B6/ES, and 1E/iPS cells. All mRNA levels are relative to those for MEF. Data shown are mean±S.D., n=4, *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). (g) Western blotting for ULK1, Beclin1, ATG5, and ATG3 in B6/MEF, B6/ES, and 1E/iPS cells. β-Actin served as the loading control. (h) Western blotting for ULK1, Beclin1, ATG5, and ATG3 during embryonic body (EB) differentiation. β-Actin served as the loading control. (i) Western blotting for LC3 during EB differentiation. β-Actin served as the loading control. Images in (a, g, h, and i) are representative of three independent experiments respectively
Autophagic flux is maintained by a core molecular machinery that is composed of an autophagy sensor system, induction complex, and an autophagosome formation system.1, 3, 24 To investigate whether enhanced autophagic flux is due to the presence of a powerful autophagic infrastructure in PSCs, we analyzed existing transcriptome data and compared the expression of core molecular machinery genes between PSCs and somatic fibroblasts using 17 ESCs, 14 iPSCs, and 12 fibroblast-independent lines.25 Approximately half of the analyzed genes were ubiquitously upregulated in both ESCs and iPSCs relative to somatic fibroblasts (Supplementary Table S1). Real-time quantitative RT-PCR was employed to verify these results (Figures 2b–f). The significant enhancement of the expression of genes involved in the autophagy initiation ULK1 complex (Ulk1, Atg13, and Atg101), supports a more efficient isolation membrane elongation and/or closure existing in PSCs (Figure 2b). Meanwhile, the expression of Pik3r4, Vps34, Becn1, Atg14, and Ambra1 in the PI3K complex also increased in PSCs, indicating that PSCs can more efficiently generate PI3P for autophagosome formation (Figure 2c). Almost all major genes involved the ATG9 cycling system, with the exception of Atg9a, Wipi1, and Vmp1 that increased expression, suggesting that PCS have a more efficient autophagosome recycling system than somatic cells (Figure 2d). This result is in agreement with the TEM assays showing that chloroquine can induce a significantly higher rate of autophagosome accumulation in PSCs than in MEFs (Figures 1c and d). Finally, expression of most core molecular machinery genes involved in both ATG12-ATG5 and LC3-PE conjugation systems was significantly upregulated in PSCs compared with somatic fibroblasts, such that PSCs likely have more efficient isolation membrane elongation and/or closure (Figures 2e and f). Accordingly, the protein levels produced by core molecular machinery genes were also increased in PSCs compared with MEFs as indicated by enhanced expression of Ulk1, Beclin1, Atg5, Atg3, and LC3 (Figure 2g and Supplementary Figure S1a). Collectively, these data suggest that PSCs are equipped with a substantial autophagy infrastructure that is maintained by enhanced expression of core molecular autophagy machinery genes.
To further confirm the correlation between core molecular machinery gene expression and autophagic flux, we established stable ESC lines harboring DOX-inducible siRNA targeting Ulk1, Beclin1, or Atg5 (the siRNA sequences are listed in Supplementary Table S3–4). We found that attenuating the expression of autophagy genes dramatically compromised autophagic flux in ESCs (Supplementary Figure S2d). These data support the assumption that high autophagic flux in PSCs is indeed maintained by a substantial autophagic infrastructure. Correspondingly, the expression of autophagy core molecular machinery genes declined together with autophagic flux during embryonic body (EB) differentiation (Figures 2h and I and Supplementary Figure S2e). Taken together, the enhanced coordinated expression of autophagy core molecular machinery genes ensures a high basal autophagic flux in ESCs.
High autophagic flux maintains ESC identity
To test the function of the high autophagic flux seen in ESCs, we first treated ESCs with different doses of 3-MA and detected their effects on pluripotency. As expected, the autophagic flux was dose-dependently inhibited by 3-MA (Supplementary Figure S3a). Colony formation and pluripotent gene expression were also inhibited by 3-MA in a dose-dependent manner (Supplementary Figures S3b–d). An EB differentiation assay showed that 3-MA treatments were associated with defective endoderm differentiation (Supplementary Figure S3e). Furthermore, genetic inhibition of autophagy by Ulk1, Beclin1, or Atg5 expression knockdown compromised self-renewal and pluripotent gene expression in ESCs (Supplementary Figures S3f–i). These data indicate that autophagy flux is closely correlated with ESC identity.
Atg3 is required for autophagosome formation.26 To further investigate how autophagy flux affects ESC identity, we isolated Atg3+/+, Atg3+/−, and Atg3−/− ESCs from embryonic day 3.5 mouse blastocysts. Immunoblotting assays showed decreased expression of Atg3 in Atg3+/− ESCs and no Atg3 expression in Atg3−/− ESCs compared with wild-type ESCs (Supplementary Figure S4a). Accordingly, wild-type ESCs have the highest autophagic flux, whereas Atg3+/− ESCs have an intermediate autophagic flux, and no canonical autophagic flux was detected in Atg3−/− ESCs (Figures 3a and d). Correspondingly, the Atg3−/− and Atg3+/− ESCs had significantly reduced self-renewal capacity and pluripotent gene expression compared with wild-type ESCs, suggesting that a high autophagy flux is essential for ESCs to maintain their stemness (Figures 3b and c and Supplementary Figure S4b). To test whether impairment of autophagic flux affects ESC differentiation, we performed teratoma and chimeric mouse formation assays using Atg3+/+, Atg3+/−, and Atg3−/− ESCs. Tapering Atg3 expression resulted in compromised teratoma formation and decreased chimeric mice formation rate, indicating the importance of high autophagic flux for ESC differentiation (Figures 3f and g).

High autophagic flux preserves ESC identity. (a) Western blotting for ATG3 and LC3 in Atg3+/+, Atg3+/−, and Atg3−/− ESCs. β-Actin served as the loading control. (b) Restore of wild-type but not V8D mutant Atg3 expression in Atg3+/−, and Atg3−/− ESCs compensated for reprogramming efficiency. (c) Quantification of AP-positive colony numbers in (b). Data shown are mean±S.D., n=3, *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). (d) Long-lived protein degradation in Atg3+/+, Atg3+/−, and Atg3−/− ESCs under EBSS starvation conditions (5 h) with or without 3-MA treatment (10 mM, 5 h). Data shown are mean±S.D., n=3, **P<0.01, ***P<0.001 (Student’s t-test). (e) qPCR for pluripotency marker genes in ATG3 rescue ESC lines. Data shown are mean±S.D., n=3, *P<0.05, **P<0.01 (Student’s t-test). (f) Statistic analysis of the weight of teratomas formed by Atg3+/+, Atg3+/−, and Atg3−/− ESCs. *P<0.05, ***P<0.001 (Student’s t-test). (g) Statistic analysis of the chimeric formation ratio of embryos (11.5 d) formed by Atg3+/+, Atg3+/−, and Atg3−/− ESCs
Mutations in the membrane-curvature-sensing domain of ATG3 result in defective autophagosome formation.27 To further confirm that the compromised self-renewal and pluripotency of Atg3+/− and Atg3−/− ESCs resulted from impaired autophagic flux, gain-of-function assays were performed by overexpressing either wild-type Atg3 or the lipidation-deficient mutant Atg3V8D in Atg3+/− and Atg3−/− ESCs (Supplementary Figure S4c). We found that gain expression of wild-type Atg3 but not Atg3V8D in Atg3+/− and Atg3−/− ESCs restored the decreased autophagic flux and mitigated the defective self-renewal and pluripotency (Figures 3b, c, and e and Supplementary Figure S4d). Taken together, these data support that a high autophagic flux guards ESC identity.
FOXO1 drives an autophagy machinery gene program to maintain high autophagic flux
We next aimed to characterize the molecular mechanisms that control coordinated increases in autophagy core machinery gene expression that maintain the high autophagic flux. Using the g: profiler tool28, 29 we found that genes encoding the autophagy core molecular machinery tend to have a coordinated gene expression program (Supplementary Figure S5a). Consistent with this result, both bioinformatic analysis of existing transcriptional data and expression analysis of PSCs and MEFs revealed that autophagy core molecular machinery genes are highly and coordinately expressed in PSCs (Figures 2b–g and Supplementary Table S1). Pattern discovery analysis of the promoter regions of 37 autophagy core molecular machinery genes identified the ‘AAACAAA’ consensus motif that is enriched in these gene promoter sets, and a total of 144 consensus motifs were identified in the 37 autophagy core molecular machinery genes (Supplementary Figures S5b–d and Supplementary Table S2). This consensus sequence is in accordance with the conserved binding domain of the forkhead family of transcription factors (FOXOs).
FOXO1 and FOXO3 are reported to trigger an autophagy core molecular machinery gene program in somatic cells.30 To determine which FOXOs coordinate the autophagy gene program in ESCs, we knocked out Foxo1 and Foxo3 by CRISPR/Cas9 (Supplementary Figure S6). Foxo1 but not Foxo3 knockout dramatically decreased autophagic flux in ESCs, indicating the involvement of FOXO1 to coordinate the autophagy gene program (Figure 4a and Supplementary Figures S7a and b). Foxo1 was highly expressed in PSCs and decreased during differentiation (Figure 4b and Supplementary Figure S5e). Foxo1 but not Foxo3 knockout compromised autophagy gene expression and attenuated starvation-induced autophagic flux increases in ESCs (Figure 4c and Supplementary Figures S7a–d). Thus, these results suggest that Foxo1 regulates expression of core molecular machinery genes that maintain autophagic flux in ESCs. To further test whether autophagy core molecular machinery genes are direct targets of FOXO1, we performed chromatin immunoprecipitation (ChIP) analysis on ESCs using an antibody specific to FOXO1. The ChIP results demonstrated that FOXO1 indeed directly binds to autophagy machinery gene promoters (Figure 4d).

FOXO1 drives an autophagy machinery gene program to maintain high autophagic flux. (a) Western blotting for LC3 in Foxo1 and Foxo3 knockout (KO) ESC lines. β-Actin served as the loading control. (b) Western blotting for FOXO1 and FOXO3 in B6/MEF, B6/ES, and 1E/iPS cells. β-Actin served as the loading control. (c) qPCR for autophagy-related genes in Foxo1 KO ESC lines. Data shown are mean±S.D., n=3, *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). (d) ChIP analysis of FOXO1 in B6/ES cells treated with rapamycin (200 nM) for 2 h. DNA co-immunoprecipitated with either anti-FOXO1 or a control pre-immune immunoglobulin (IgG) were quantitated by qPCR and normalized to input. Representative results from three independent experiments are shown. (e) Reacquisition of wild-type but not mutant Foxo1 expression in Foxo1−/− cells compensated for reprogramming efficiency. (f) Quantification of AP-positive colonies from (e). Data shown are mean±S.D., n=3, *P<0.05, **P<0.01 (Student’s t-test). (g) qPCR for pluripotency marker genes. Data shown are mean±S.D., n=3, *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). Images in (a and b) are representative of three independent experiments respectively
FOXO1 is a forkhead transcription factor that carries a winged helix DNA-binding domain known as a forkhead box.30 To further confirm that the impaired autophagy machinery gene expression in Foxo1−/− ESCs is directly caused by the loss of Foxo1 transcriptional activity, a gain-of-function assay was performed by introducing a Foxo1 expression vector into Foxo1−/− ESCs. We established stable Foxo1−/− ESC lines carrying an empty vector, wild-type Foxo1, or a DNA-binding deficient Foxo1DB mutant (Supplementary Figure S7e).31 As expected, autophagy gene expression was restored only in Foxo1−/− stable ESC lines carrying wild-type Foxo1 but not those with the empty vector or the Foxo1DB mutant (Supplementary Figures S7f and g). These results support that the autophagy core molecular machinery genes are directly regulated by Foxo1. Correspondingly, the decreased autophagic flux was rescued in Foxo1−/− stable ESCs expressing wild-type Foxo1 (Supplementary Figure S7h). Collectively, these data demonstrate that FOXO1 drives an autophagy core molecular machinery gene program to maintain the high autophagic flux seen in ESCs.
FOXO1 regulates self-renewal and pluripotency of ESCs
We next investigated whether FOXO1 regulates ESC identity. Both Foxo1 and Foxo3 were highly expressed in ESCs, but loss of FOXO1 but not FOXO3 in ESCs significantly decreased the clonogenic survival rate, suggesting that Foxo1 is pivotal for ESC self-renewal (Figure 4b and Supplementary Figures S8a and b). Expression of pluripotent genes in Foxo1−/− but not Foxo3−/− ESCs was also significantly decreased, indicating that the loss of FOXO1 compromised ESC pluripotency (Supplementary Figures S8c and d). Furthermore, Foxo1−/− but not Foxo3−/− ESCs had abnormal EB differentiation, characterized by defective expression of certain endodermic and mesodermic marker genes that is consistent with that of Atg3−/− ESCs, and defective teratoma formation (Supplementary Figures S8e and f).
To further confirm that the compromised self-renewal ability and pluripotency of Foxo1−/− ESC was directly caused by decreases in Foxo1-mediated autophagic flux, we next performed gain-of-function assays. We found that the clonogenic formation rate and expression of pluripotent genes of Foxo1−/− ESCs could be restored only by wild-type Foxo1 but not empty vector or the Foxo1DB mutant (Figures 4e–g and Supplementary Figure S8g). Together, these data indicate that FOXO1 regulates self-renewal and pluripotency by coordinating the autophagy core molecular machinery gene program that maintains high autophagic flux.
Discussion
ESCs are immortal in culture and have a high proliferation rate with a short G1 phase. Proliferating cells require not only energy (ATP) and reducing cofactors (NADH), but also building blocks to replicate macromolecular components during each cell cycle passage.32, 33, 34 Recent studies showed that increased proteasome activity is an intrinsic characteristic of human ESCs and proposed that human ESCs use enhanced proteasome activity to maintain an intact proteome for ESC self-renewal and pluripotency.35 In contrast, we demonstrate that ESCs harness high autophagic flux to guard their self-renewal and pluripotency capacity. FOXO1 is a master regulator that coordinates the high expression of autophagy core molecular machinery genes in ESCs to maintain their high autophagic flux. We propose that high autophagic flux driven by FOXO1 protects stemness of ESC through efficient generation of molecular building blocks for use in rapid cell proliferation and/or quick removal of damaged organelles and aggregated proteins that avoid oxidative damage to the genome. Consistent with this hypothesis, Atg5 deletion has been shown to compromise degradation of long-lived proteins,36 and lack of Atg3-dependent autophagy removal results in accumulation of abnormal mitochondria, increased ROS production, and defective ATP generation in ESCs.18
Although accurate regulation of autophagy by nutrient-responsive kinases is well defined, long-term regulation of autophagy at transcriptional level is poorly understood.2, 3, 37 Recent studies suggest that transcriptional regulation of autophagic genes is one of the most important molecular mechanisms for functional regulation of autophagy in differentiated cells. In support of this assumption, some studies have identified several transcription factors like TFEB, GATA1, and FOXO3 as positive and ZKSCAN3 as negative master regulators to coordinately modulate a panel of autophagic gene expression in response to distinct stresses.38, 39, 40, 41 Meanwhile, other studies have found that activation of nuclear receptor farnesoid X receptor (FXR) can inhibit autophagy through downregulation of autophagic gene expression.42, 43 These studies underline the functional importance of transcriptional regulation of autophagy. In contrast, we have identified that enhanced autophagy molecular machinery, which is transcriptionally regulated by FOXO1, endows ESCs with a powerful autophagic flux to maintain their function integrity. In this point, the finding highlights the transcriptional regulation of autophagy as a critical molecular mechanism to guard cellular identity in pluripotent stem cells.
FOXO transcription factors function as master regulators to control cellular homeostasis in multiple biological processes, whose activity can be inhibited by insulin and growth factor signaling.30 The mammalian FOXO family, which comprises four members FOXO1, FOXO3, FOXO4, and FOXO6, shows differential but overlapping expression throughout the organism, indicating their functional specificity and diversity.44 In muscle and cardiomyocyte cells, FOXO3 is identified to regulate autophagy in response to starvation through directly upregulating expression of autophagic genes.38, 45, 46 In adult stem cells like hematopoietic stem cell (HSC), FOXO3 has also been found to direct a protective autophagic gene expression program for HSC self-renewal and maintenance of HSC pool.9 In contrast, we have identified that FOXO1 drives a high autophagic flux by coordinating autophagic gene program to maintain ESC identity. These data support the hypothesis that different FOXO family members bind to promoters of target genes with distinct affinities to facilitate their subtle functional division in differential biology process.44 Most interestingly, a recent study found FOXO1 can regulate ESC pluripotency by direct targeting Oct4 and Sox2.47 These results suggest that FOXO1 regulates ESC identity by directly targeting both pluripotent genes and autophagic genes, highlighting a novel elaborate autophagy regulation mechanism in ESCs.
In conclusion, our findings define a previously uncharacterized role and mechanism of autophagy in ESC identity regulation, and offer new insights into the current understanding of pluripotency regulation. In addition, this discovery subverts the traditional hypothesis that autophagy is required for nondividing or differentiated cells rather than rapidly proliferating cells, and highlights a pivotal function of autophagy in fast proliferation ESCs.
Materials and methods
Mice, cell culture, and reagents
Atg3+/− mice were purchased from Riken (RBRC02761, Tsukuba, Japan). All protocols used in experiments involving animals were approved by the institutional animal care committee. B6/MEFs were isolated from embryonic day 13.5 (E13.5 d) mice and used within three generations. MEF and HEK293T cells were maintained in DMEM high glucose with 10% fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, and 1% penicillin/streptomycin. B6 Atg3+/+, Atg3+/−, and Atg3−/− ES cell lines were isolated from E3.5 d mice and cultured on feeder layers for 5 d and then routinely passaged. All ESCs and iPSCs were maintained in knockout DMEM with 15% fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1% penicillin/streptomycin, 0.055 mM β-mercaptoethanol, and 1000 U/ml leukemia inhibitory factor (Millipore, Darmstadt, Germany) as previously reported.48, 49 All cell culture reagents were purchased from Gibco (Carlsbad, CA, USA) unless indicated. Rapamycin, 3-MA, Baf-A1, chloroquine (CQ), and doxycycline (Dox) were purchased from Sigma (St. Louis, MO, USA).
Generation of stable ESC lines
Tetracycline repressor protein (TetR) and full-length mouse ATG3 and FOXO1 cDNA were cloned into pCDH-CAG-RFP lentiviral expression vectors, and various mutations were generated by overlap-PCR using the primers listed in Supplementary Table S3-2. Lentiviral particles were produced by calcium phosphate-mediated cotransfection of HEK293T cells with psPAX2 and pMD2.G plasmids. Supernatants containing the lentiviral particles were collected 48 h after transfections. Mouse ESCs were infected by incubation with lentivirus-containing supernatants for 24 h. After trypsinization, infected ESCs were subcultured at a low density on CF1 MEF feeder layers. After culturing for 6 days, RFP-positive single colonies were picked and cultured normally.
Generation of Foxo1 and Foxo3 knockout ESCs
Foxo1 and Foxo3 knockout ESC lines were generated using a CRISPR-Cas9 system. Briefly, optimal Foxo1 and Foxo3 sgRNA oligonucleotides were cloned into the px330 vector, and 30 μg sgRNA plasmids were electrophoretically transferred to B6/ES cells. After transformation, ESCs were plated at a low density on CF1 MEF feeder layers. Single colonies were picked, cultured, and genotyped by RFLP. Additional sequencing and western blotting was performed to confirm the presence of mutations in the colonies.
Western blotting
Cells were lysed on ice in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 5 mM EGTA, 2 mM EDTA, 10 mM NaF) for 30 min and a protease inhibitor cocktail (04693116001, Roche, Mannheim, Germany) and 1 mM PMSF (ST506, Beyotime, Shanghai, China) was added. The lysates were mixed with loading buffer, boiled, centrifuged, and subjected to SDS-PAGE electrophoresis before transfer to PVDF membranes (Millipore). After blocking with 5% non-fat milk in TBST (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% Tween-20), the membrane was incubated with the following primary antibodies: anti-LC3B (L7543, Sigma), anti-mTOR (2972, Cell Signaling Technology, Danvers, MA, USA), anti-p-mTOR (Ser2448) (2971, Cell Signaling Technology), anti-S6K (2708, Cell Signaling Technology), anti-p-S6K (Thr389) (9205, Cell Signaling Technology), anti-ULK1 (A7481, Sigma), anti-p-ULK1 (Ser757) (6888, Cell Signaling Technology), anti-Beclin1 (sc-11427, Santa Cruz, CA, USA), anti-ATG3 (ab108251, Abcam, Cambridge, MA, USA), anti-ATG5 (ab108327, Abcam), anti-p62 (ab56416, Abcam, Cambridge, MA, USA), anti-FOXO1 (2890, Cell Signaling Technology), anti-FOXO3 (2497, Cell Signaling Technology), and anti-β-actin (A5441, Sigma). The secondary antibodies used were goat anti-rabbit (sc-2030, Santa Cruz) and goat anti-mouse (sc-2031, Santa Cruz) IgG antibodies. Finally, protein expression was detected by Luminata Forte Western HRP Substrate (WBLUF0100, Millipore), and protein bands were quantified using Image J Software (Bethesda, MD, USA).
Transmission electron microscopy
For TEM, MEFs and PSCs were fixed with 2.5% glutaraldehyde at 4 °C for at least 2 h followed by postfixation in 2% osmium tetroxide (OsO4) for 2 h. After a graded-ethanol serial dehydration, the samples were embedded in epoxy resins. Ultrathin sections (70 nm) were prepared with a diamond knife on an ultramicrotome (Leica EM UC6, Vienna, Austria) and collected on copper grids. Sections were then stained with uranyl acetate and lead citrate, and observed with a Hitachi H-7650B TEM (Hitachi, Tokyo, Japan) operating at 80 kV.
Long-lived protein degradation assay
MEFs and PSCs were labeled for 24 h with 0.2 μCi/ml L-[14C]valine. The cells were then washed three times with PBS to remove unincorporated radioisotopes, and incubated with 10 mM unlabeled L-valine in complete growth medium for 1 h (prechase period). After the prechase step, cells were incubated for the designated times in either complete growth medium or EBSS containing 10 mM unlabeled valine with or without 10 mM 3-MA (chase period). After the chase step, proteins from the medium and cells were precipitated separately with 10% (w/v) TCA (overnight, 4 °C). The TCA-precipitated fractions were centrifuged (600 × g, 20 min, RT) and redissolved in 1 ml 0.2 N NaOH. Finally, the radioactivity was determined by liquid scintillation counting, and the rate of degradation of long-lived proteins was calculated by determining the ratio between TCA-precipitated radioactivity recovered from the medium and TCA-precipitated radioactivity from the medium and cells.
Real-time PCR
Total RNA was extracted from samples with an RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Approximately 1–2 μg total RNA was reverse-transcribed into cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). qPCR was performed with GoTaq qPCR Master Mix (Promega, Madison, WI, USA) and a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA); all samples were analyzed in duplicate and normalized to β-actin. The primers used are listed in Supplementary Table S3–1.
Clonogenic survival assay
A clonogenic survival assay was performed as previously described.18 In brief, mouse ESCs were trypsinized into single cells and seeded into six-well plates with a thin feeder layer at a density of 1 × 103 per well for further culture. After 7 days of culture, ES colonies were stained with a BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. AP-positive ES colonies in each well were then counted and photographed.
EB formation assay
Mouse ESCs were dissociated as clusters with 0.05% trypsin-EDTA, seeded in bacterial-grade 60 mm dishes, and cultured with MEF culture medium without LIF. The medium was changed every other day. EBs were collected after 0, 3, 6, or 9 days and then used for further analysis.
Teratoma formation assay
Trypsinized mouse ESCs (1–2 × 106) were subcutaneously injected into 6-week-old NOD SCID mice (Vital River, Beijing, china). After 4 to 6 weeks, the resulting tumors were surgically removed, weighed, and fixed in 4% paraformaldehyde overnight and embedded in paraffin. Sections were prepared and stained with hematoxylin and eosin for histological examination as previously reported.50
ChIP assay
ChIP assays were performed using a Magna ChIP A/G ChIP Kit (17–10085, Millipore) according to the manufacturer’s protocol. Briefly, 1 × 107 B6/ES cells were fixed with 1% paraformaldehyde, washed, resuspended in lysis buffer, and sonicated. Lysates containing soluble chromatin were prepared and incubated with antibodies specific to FOXO1 (ab39670, Abcam) and control rabbit IgG (sc-2027, Santa Cruz). After the protein/DNA complexes were eluted, the crosslinking was reversed and the purified. The specific DNA fragments were quantitated by qRT-PCR and normalized to input. ChIP PCR analysis primer sets were designed to cover the FOXO binding region of the promoter region in selected autophagy-related genes and are listed in Supplementary Table S3-3.
Cell proliferation and apoptosis assay
CFSE-labeled live cells were used to quantify cell proliferation in vitro. Briefly, 1 × 106 ES cells were incubated with 5 μM CFSE (21888, Sigma) for 15 min at 37 °C, and then washed three times. After CFSE labeling, 2 × 105 cells were seeded into a 6-well plate for further culture. Cells were then harvested at the indicated time points and analyzed by flow cytometry. For apoptosis detection, an Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Invitrogen) was used according to the manufacturer’s instructions. Briefly, cells were washed with PBS and resuspended in binding buffer. The cells were then incubated with annexin-V-FITC and propidium iodide (PI) for 20 min at RT, and subsequently analyzed by flow cytometry. Cells that were annexin-V negative and PI negative were counted as viable cells, and those that were annexin-V positive and PI negative were counted as apoptotic cells.
Expression correlation and sequence alignment analysis
Expression correlation analysis was performed using the g: profiler tool as previously described.28, 29 Sequence alignment analysis was performed to identify transcription factor binding sites in the regulatory regions of autophagy-related genes as previously described.51
Statistical analysis
All data were derived from multiple independent experiments rather than technical replicates. Most were presented as mean±S.D. Each exact n value is stated in the corresponding figure legend. Statistical significance was calculated using the two-tailed Student’s t-test between the indicated groups, and P-values of <0.05 were considered statistically significant (*P<0.05, **P<0.01, ***P<0.001).
References
Klionsky DJ . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8: 931–937.
He C, Klionsky DJ . Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43: 67–93.
Mizushima N, Yoshimori T, Ohsumi Y . The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27: 107–132.
Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ . Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995; 270: 2320–2326.
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20: 1981–1991.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20: 1992–2003.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2013; 13: 132–141.
Mizushima N, Levine B . Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12: 823–830.
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013; 494: 323–327.
Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E et al. Autophagy maintains stemness by preventing senescence. Nature 2016; 529: 37–42.
Cao J, Li X, Lu X, Zhang C, Yu H, Zhao T . Cells derived from iPSC can be immunogenic - yes or no? Protein Cell 2014; 5: 1–3.
Liu K, Song Y, Yu H, Zhao T . Understanding the roadmaps to induced pluripotency. Cell Death Dis 2014; 5: e1232.
Gao R, Liu XY, Gao SR . Progress in understanding epigenetic remodeling during induced pluripotency. Sci Bull 2015; 60: 1713–1721.
Li JS . Stem cell, basis and application. Sci Bull 2015; 60: 1711–1712.
Wang S, Xia P, Ye B, Huang G, Liu J, Fan Z . Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 2013; 13: 617–625.
Ma T, Li J, Xu Y, Yu C, Xu T, Wang H et al. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol 2015; 17: 1379–1387.
Wu Y, Li Y, Zhang H, Huang Y, Zhao P, Tang Y et al. Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat Cell Biol 2015; 17: 715–725.
Liu K, Zhao Q, Liu P, Cao J, Gong J, Wang C et al. ATG3-dependent autophagy mediates mitochondrial homeostasis in pluripotency acquirement and maintenance. Autophagy 2016; 12: 2000–2008 1-9.
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–326.
Vabulas RM, Hartl FU . Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 2005; 310: 1960–1963.
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544.
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465: 942–946.
Alers S, Loffler AS, Wesselborg S, Stork B . Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32: 2–11.
Yang Z, Klionsky DJ . Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22: 124–131.
Zhu S, Cao J, Sun H, Liu K, Li Y, Zhao T . p18 inhibits reprogramming through inactivation of Cdk4/6. Sci Rep 2016; 6: 31085.
Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 2008; 19: 4762–4775.
Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol 2014; 16: 415–424.
Reimand J, Kull M, Peterson H, Hansen J, Vilo J . g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 007; 35: W193–W200.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325: 473–477.
Webb AE, Brunet A . FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 2014; 39: 159–169.
Arriola DJ, Mayo SL, Skarra DV, Benson CA, Thackray VG . FOXO1 transcription factor inhibits luteinizing hormone beta gene expression in pituitary gonadotrope cells. J Biol Chem 2012; 287: 33424–33435.
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Folmes CD, Dzeja PP, Nelson TJ, Terzic A . Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012; 11: 596–606.
Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA . Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012; 11: 589–595.
Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012; 489: 304–308.
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152: 657–668.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–141.
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6: 458–471.
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332: 1429–1433.
Kang YA, Sanalkumar R, O'Geen H, Linnemann AK, Chang CJ, Bouhassira EE et al. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 2012; 32: 226–239.
Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell 2013; 50: 16–28.
Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014; 516: 112–115.
Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516: 108–111.
Salih DA, Brunet A . FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008; 20: 126–136.
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6: 472–483.
Sengupta A, Molkentin JD, Yutzey KE . FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 2009; 284: 28319–28331.
Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, Su J et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol 2011; 13: 1092–1099.
Wang L, Cao J, Wang Y, Lan T, Liu L, Wang W et al. Immunogenicity and functional evaluation of iPSC-derived organs for transplantation. Cell Discov 2015; 1: 15015.
Zhang C, Cao J, Li X, Xu H, Wang W, Wang L et al. Treatment of multiple sclerosis by transplantation of neural stem cells derived from induced pluripotent stem cells. Sci China Life Sci 2016; 59: 950–957.
Zhao T, Zhang ZN, Rong Z, Xu Y . Immunogenicity of induced pluripotent stem cells. Nature 2011; 474: 212–215.
Lubke T, Lobel P, Sleat DE . Proteomics of the lysosome. Biochim Biophys Acta 2009; 1793: 625–635.
Acknowledgements
We sincerely thank Dr. Dangsheng Li for his constructive suggestions and Dr. Weiguo Zhu for his generous plasmids. This work was supported by grants from the China National Basic Research Program (2013CB966901, 2012CBA01303), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA01040108), the National Natural Science Foundation of China Program (31271592, 31570995, 31621004), and National Thousand Young Talents Program to TZ.
Author contributions
PL and TZ designed experiments, analyzed data, and wrote the paper; PL, KL, HG, JG, WW, YZ, QZ, and JC performed experiments under the overall coordination of TZ. FG, CH, QC, WL, JJ, BH, QZ, and TZ supervised experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Liu, P., Liu, K., Gu, H. et al. High autophagic flux guards ESC identity through coordinating autophagy machinery gene program by FOXO1. Cell Death Differ 24, 1672–1680 (2017). https://doi.org/10.1038/cdd.2017.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.90
This article is cited by
-
Forkhead box O proteins: steering the course of stem cell fate
Cell Regeneration (2024)
-
Dynamic Autophagy Map in Mouse Female Germ Cells Throughout the Fetal to Postnatal Life
Reproductive Sciences (2023)
-
BNIP3 (BCL2 interacting protein 3) regulates pluripotency by modulating mitochondrial homeostasis via mitophagy
Cell Death & Disease (2022)
-
Autophagy-related DjAtg1-1 plays critical role in planarian regeneration by regulating proliferation and cell death
Cell and Tissue Research (2022)
-
Dissection of two routes to naïve pluripotency using different kinase inhibitors
Nature Communications (2021)
