
Abstract
CX3CR1, a G protein-coupled receptor solely expressed by microglia in the brain, has been repeatedly reported to be associated with neurodevelopmental disorders including schizophrenia (SCZ) and autism spectrum disorders (ASD) in transcriptomic and animal studies but not in genetic studies. To address the impacts of variants in CX3CR1 on neurodevelopmental disorders, we conducted coding exon-targeted resequencing of CX3CR1 in 370 Japanese SCZ and 192 ASD patients using next-generation sequencing technology, followed by a genetic association study in a sample comprising 7054 unrelated individuals (2653 SCZ, 574 ASD and 3827 controls). We then performed in silico three-dimensional (3D) structural modeling and in vivo disruption of Akt phosphorylation to determine the impact of the detected variant on CX3CR1-dependent signal transduction. We detected a statistically significant association between the variant Ala55Thr in CX3CR1 with SCZ and ASD phenotypes (odds ratio=8.3, P=0.020). A 3D structural model indicated that Ala55Thr could destabilize the conformation of the CX3CR1 helix 8 and affect its interaction with a heterotrimeric G protein. In vitro functional analysis showed that the CX3CR1-Ala55Thr mutation inhibited cell signaling induced by fractalkine, the ligand for CX3CR1. The combined data suggested that the variant Ala55Thr in CX3CR1 might result in the disruption of CX3CR1 signaling. Our results strengthen the association between microglia-specific genes and neurodevelopmental disorders.
Similar content being viewed by others


Introduction
Both schizophrenia (SCZ) and autism spectrum disorders (ASD) are highly polygenic neurodevelopmental disorders. SCZ and ASD share etiological, clinical and biological features.1, 2 The prevalence and recurrence risks to relatives are nearly identical, and heritability is estimated to be a minimum of 80% for each disorder.3 Genetic contributions have played a major role in the etiology of both disorder; however, these genetic components are still unclear. As thousands of trait- and disease-associated common genetic variants of small effect may explain less than half of the total variation responsible for increased risk of developing either disorder,4, 5, 6 a significant excess of rare, disruptive variants (with a frequency <1%) have provided additional genetic risk of these complex disorders or trait variability.7, 8, 9, 10, 11 In particular, many genes with rare variants that are common to both SCZ and ASD are related to synapse morphogenesis.12, 13, 14, 15
Recent evidence implicates synaptic formation as an important factor in the pathogenesis of SCZ and ASD.12, 16 It is becoming clear that microglia, which are the resident macrophages and phagocytes of the central nerve system, contribute to major aspects of the structural shaping and functional modulation of the connectivity of the developing and healthy brain.17, 18, 19, 20, 21 Aberrant functions of microglia have potential implication for SCZ and ASD.22, 23, 24, 25 Altered gene expression of microglia has been found in the brains of SCZ26, 27 and ASD patients.28 Transcriptomic studies of post-mortem brains have demonstrated dysregulation of microglial gene expression in SCZ29 and ASD.30 Microglial synapse morphogenesis is dependent on the receptor CX3C chemokine receptor 1 (CX3CR1, also known as fractalkine receptor, OMIM: 601470), which is solely expressed by microglia in the brain19 and has been proposed as a key mediator of neuron–microglia interactions.31 Mice lacking Cx3cr1 exhibit deficits in synaptic pruning19, 20, 32, 33, 34 as well as in social interaction and increased repetitive-behavior phenotypes, which is considered to be the core symptoms of ASD.19, 34 These findings strongly support the notion that CX3CR1 is a plausible candidate risk gene for SCZ and/or ASD.
To date, two CX3CR1 polymorphism, Var249Ile (rs3732379) and Thr280Met (rs3732378), have been reported to affect the activity of the CX3CR1 protein. These polymorphisms are associated with several neuroinflammatory disorders such as HIV-1 infection,35 multiple sclerosis,36 amytrophic lateral sclerosis,37 age-related macular degeneration38 and coronary atherosclerosis.39 However, the effects of these common variants on disease pathogenesis are quite small.35, 36, 37, 38 There has been no published report of causal genetic variants of CX3CR1 in cases with neurodevelopmental disorders including SCZ and ASD. We therefore hypothesized that a rare variant of the microglia-specific CX3CR1 gene would contribute to some pathophysiological mechanisms of these neurodevelopmental disorders.40 To increase statistical power and detect shared risk, we combined SCZ and ASD samples in a study cohort.5, 15
We then performed in silico three-dimensional (3D) structural modeling and in vivo disruption of Akt phosphorylation to determine the impact of the detected variant on CX3CR1-dependent signal transduction based on the following: (i) CX3CR1 is a seven transmembrane domain G protein-coupled receptor (GPCR), the signaling systems of which are involved in many disease and major therapeutic targets.41 (ii) The chemokine fractalkine, the sole ligand for CX3CR1, is known to activate the Akt signaling pathways through CX3CR1.42, 43, 44, 45, 46
In this study, we present the genetic association of a variant in CX3CR1 with SCZ and ASD, followed by in silico 3D structural modeling of the predicted conformational change of the variant receptor and its in vivo disruption of the Akt signaling. Our findings provide the genetic evidence of the association of a microglia-specific gene with neurodevelopmental disorders.
Materials and methods
Study samples
Two independent Japanese sample groups were used in this study. The targeted-resequencing discovery cohort comprised 370 SCZ (mean age±s.d., 49.7±14.8 years; 53% male) and 192 ASD (mean age±s.d.=16.3±8.4 years; 77.6% male). For the genetic association analysis, the case–control sample set comprised 2283 SCZ (48.1±23.8 years; 41.7% male), 382 ASD (19.6±10.7 years; 77.8% male) and 3827 control subjects (43.3±14.5 years; 41.6% male). All subjects were unrelated, living on the mainland of Japan, and self-identified as Japanese. All patients fulfilled the criteria listed in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) for SCZ or ASD. Healthy control subjects were selected from the general population and had no history of mental disorders based on questionnaire responses from the subjects themselves during the sample inclusion step. The study was explained to each participant and/or their parents both verbally and in writing. Written informed consent was obtained from the participants and from the parents for patients under 20 years old. All procedures performed in this study involving human participants were approved by the Ethics Committee of the Nagoya University Graduate School of Medicine. They were conducted in accordance with the Helsinki Declaration of 1975 and its later amendments or comparable ethical standards.
Sample preparation
Genomic DNA was extracted from peripheral blood or saliva from each SCZ, ASD and control participant using the QIAamp DNA Blood Kit or Tissue Kit (Qiagen, Hilden, Germany). The quantity of extracted DNA was estimated using the Qubit dsDNA BR Assay Kit (Life Technologies, Carlsbad, CA, USA) on a Qubit 2.0 Fluorometer (Life Technologies) following the manufacturer’s recommended protocol.
Library preparation and resequencing
We used the next-generation sequencing technology of the Ion Torrent PGMto resequence the CX3CR1 coding regions (Ensembl Transcript ID: ENST00000399220, NCBI reference sequence NM_001337.3, NP_001328.1; 355 amino acids) via the protocols described in the Ion AmpliSeq Library Preparation User Guide (Thermo Fisher Scientific, Waltham, MA, USA, Rev.5; MAN0006735), the Ion PGM Template OT2 200 Kit (Thermo Fisher Scientific, Rev. 5; MAN0007220) and the Ion PGM Sequencing 200 Kit (Thermo Fisher Scientific, Rev. 3; MAN0007273). Custom amplification primers were designed to cover coding exons and flanking intron regions of the selected genes with Ion AmpliSeq Designer (Thermo Fisher Scientific). Sample amplification and equalization were achieved using Ion AmpliSeq Library Kits 2.0 and the Ion Library Equalizer Kit, respectively (Thermo Fisher Scientific). Amplified sequences were ligated with Ion Xpress Barcode Adapters (Thermo Fisher Scientific). Emulsion PCR and subsequent enrichment were performed using the Ion OneTouch Template Kit v2.0 on Ion OneTouch 2 and Ion OneTouch ES, respectively (Thermo Fisher Scientific).
Data analysis
Sequence reads were run through a data analysis pipeline of the Ion Torrent platform-specific pipeline software, Torrent Suite version 4.4 (Life Technologies) to generate sequence reads filtered according to the pipeline software quality-controls and to remove poor signal reads. Reads assembly and variant identification were performed using the Ingenuity Variant Analysis software (http://www.ingenuity.com/variants) from Ingenuity Systems using Fastq files containing sequence reads and the Ion Ampliseq Designer BED file software to map the amplicons with default parameters (call quality >20 and read depth >10).
Candidate variants were defined as exonic or splice-site variants with allele frequencies of ⩽1% in the following three public exome databases: dbSNP Build 149 (http://www.ncbi.nlm.nih.gov/projects/SNP/); the 1000 Genomes Project (http://www.1000genomes.org); and the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org). We then examined two databases as a reference for Japanese controls: the Human Genetic Variation Database (HGVD) (http://www.genome.med.kyoto-u.ac.jp/SnpDB/) and the integrative Japanese Genome Variation Database (iJGVD) (https://ijgvd.megabank.tohoku.ac.jp).47 Prediction of significance was performed using PolyPhen-2 (last accessed Feb 2017; http://genetics.bwh.harvard.edu/pph2/)48 and MutationTaster (last accessed Feb 2017; http://www.mutationtaster.org).49 Additional clinical variant annotations were obtained from NCBI ClinVar (last accessed Feb 2017; http://www.ncbi.nlm.nih.gov/clinvar/)50 and DECIPHER v9.12 (last accessed Feb 2017; https://decipher.sanger.ac.uk).51 Localization of a protein domain was based on the human protein reference database (http://www.hprd.org/index.html) (HPRD). When available, parents were sequenced to determine inheritance patterns. All candidate variants were confirmed by Sanger sequencing with the ABI 3130xl Genetic Analyzer (Life Technologies) with standard methods. Sequence analysis software version 6.0 (Applied Biosystems, Foster City, CA, USA) was used to analyze all sequence data. Primer sequences for validating each variant are available in Supplementary Table S3.
Genetic association analysis
The effective sample size and statistical power was computed using the web browser program, Genetic Power Calculator, developed by Purcell et al. (http://pngu.mgh.harvard.edu/~purcell/gpc/).52 An ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) and TaqMan assays with custom probes were used to genotype a putative deleterious variant. Custom probe sequences are indicated in Supplementary Table S4. Each 384-microtiter plate contained two non-template controls and two samples with the variant. The reactions and data analysis were performed using Genotyping Master Mix and Sequence Detection Systems, respectively, according to standard protocols (Applied Biosystems). Differences in genotype distribution between cases and controls were tested with one-sided, Fisher’s exact tests.
Phenotypic analysis
The clinical features of patients with the variant that was possibly associated with SCZ and ASD phenotypes based on genetic association analysis were examined retrospectively from medical records. All comorbidities were diagnosed by experienced psychiatrists according to DSM-5 criteria.
Modeling of the 3D structure of CX3CR1-Ala55Thr
A 3D structure of CX3CR1 could be modeled using a homology modeling technique, because many 3D structures of members of the GPCR family have been solved recently. The 3D structure of CCR2 isoform B53 was chosen as the template for this modeling (PDBcode: 5t1a), as this structure was the closest homolog to CX3CR1 that was found by the BLAST program in the 3D structural database.54 The sequence identity between CX3CR1 and CCR2 isoform B is about 46%. The template structure and the BLAST alignment were downloaded from the HOMCOS server,55 and manually modified to remove the region of T4 lysozome in the structure. The program MODELLER 9.16 was used for the modeling.56 Bound structures of membrane, fractalkine and heterotrimeric G protein were modeled by 3D superimpositions. 3D structures of GPCR in complexes were superimposed on the CX3CR1 model using the program MATRAS,57 and bound molecules in the complexes were also transformed with their GPCRs. The membrane model used was taken from a molecular dynamics study of GPCRs.58 The fractalkine structure was obtained from a complex of fractalkine with a viral GPCR (PDBcode: 4xt1).59 The structure of a heterotrimeric G protein (Gα, Gβ, Gγ) was taken from a G protein complex with the β2 adrenergic receptor (PDBcode: 3sn6).60
Functional analysis of CX3CR1-Ala55Thr; effect on Akt phosphorylation-mediated signaling
The cDNA of human CX3CR1 was amplified from the IMAGE clone 5216452, and was inserted into the pmCherry vector (Clontech, Palo Alto, CA, USA) using Bgl2 and EcoR1. For the construction of pmCherry-Cx3cr1 Ala55Thr, site-specific mutagenesis was carried out using the KOD-Plus-Mutagenesis Kit (TOYOBO, Shiga, Japan) with pmCherry-Cx3cr1 wild-type (WT) as a template. The following primers were used for mutagenesis: (1) forward 5′-GTGTTTACCCTCACCAACAGCAAGA-3′ and (2) reverse 5′-TACCAACAAATTTCCCACCAGGCCA-3′.
Cells of the human embryonic kidney cell line, HEK293, were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and 50 U penicillin–streptomycin at 37 °C with 5% CO2 inside a humidified incubator. Cells were transiently transfected with the pmCherry-Cx3cr1 WT or Ala55Thr plasmid by using Lipofectamine 2000 (Invitrogen), and cultured for 36 h. Then, the cells were fixed with 4% paraformaldehyde for 30 min and washed with PBS. Subsequently, the cells were mounted with a fluorescence mounting medium (DakoCytomation, Glostrup, Denmark). Images were acquired on a microscope (BX51; Olympus, Tokyo, Japan) equipped with a sCMOS camera (Zyla; Andor Technology, Belfast, UK).
For western blotting, the cells were transfected with the pmCherry-Cx3cr1 WT or Ala55Thr plasmid, cultured for 36 h, and then stimulated with fractalkine (R&D systems, Minneapolis, MN, USA) for 15 min. The cell lysates were boiled in sample buffer for 5 min. The proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membrane was blocked with 5% non-fat dry milk in PBS containing 0.05% Tween-20 (PBS-T) and incubated for 1 h at room temperature with the primary antibody diluted in PBS-T containing 1% non-fat dry milk. After washing in PBS-T, the membrane was incubated with an HRP-conjugated anti-rabbit IgG antibody (Cell Signaling Technology, Danvers, MA, USA). The ECL chemiluminescence system (GE Healthcare, Waukesha, WI, USA) was used for signal detection. Signals were detected using the Amersham Imager 600 (GE Healthcare), and were quantified using Image J software (NIH, Bethesda, MD, USA, in public domain software).
Results
Screening of variation in CX3CR1 coding exons
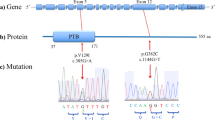
Nucleotide sequence data have been submitted to the DNA Data Bank of Japan (DDBJ) databases (http://www.ddbj.nig.ac.jp) under the accession number DRA004490. We identified three rare missense heterozygous variants within CX3CR1 coding regions in genomic DNA isolated from Japanese SCZ or ASD patient samples (n=562). All of the single nucleotide variants (SNVs) were validated by Sanger sequencing (Figure 1; Table 1). Nonsense variants, frameshift variants and splicing-site variants were not found. Genomic DNA of the parents was available for three of seven subjects carrying these three rare missense variants. In these three pedigrees, the variants were found to be transmitted from a parent (Supplementary Figure S1). None of SNVs detected in our study were registered in ClinVar nor in the DECIPHER database.

Genomic locations of the variants in CX3CR1 detected in this study. Note: Localization of a protein domain is based on the human protein reference database (http://www.hprd.org/index.html) (HPRD). Genomic position is based on NCBI builds GRCh38 (Ensembl Transcript ID ENST000000399220). Amino acid position is based on the NCBI reference sequences NP_001328.1 and DDBJ DRA004490. UTR, untranslated regions.
Genetic association of CX3CR1-Ala55Thr with SCZ and ASD
Of these three SNVs, we focused on the Ala55Thr variant because it was detected in two unrelated cases in a 562-patient cohort and was present in only one of the 60,360 Exome Aggregation Consortium (ExAC) cohort (http://exac.broadinstitute.org) (Table 1), whereas the Gly112 Ala variant was registered in Japanese databases: the HGVD (http://www.genome.med.kyoto-u.ac.jp/SnpDB/) and the integrative Japanese Genome Variation Database (iJGVD) (https://ijgvd.megabank.tohoku.ac.jp)60 with approximately the same frequency as that of our cohort, and the Met138Ile variant was found in multiple Asian individuals in ExAC (Table 1).
The effective sample size and statistical power was computed using the web browser program, Genetic Power Calculator (http://pngu.mgh.harvard.edu/~purcell/gpc/).61 For our sample set of cases (n=3227) and controls (n=3827), we computed a statistical power of >80% using the following parameters: disease prevalence of 0.01; observed rare-allele frequency of 0.0018; odds ratio for dominant effect of ⩾2.3; and type I error rate of 0.05. Differences in genotype distribution between cases and controls were tested with one-sided, Fisher’s exact tests. A significant association between neurodevelopmental disorders (SCZ and ASD) and Ala55Thr (Odds ratio=8.3, P=0.020) was observed (Table 2; Supplementary Table S1). The clinical features of the five SCZ and two ASD cases, and the one healthy control case with CX3CR1-Ala55Thr are shown in the Supplementary Table S2.
Modeling of the 3D structure of CX3CR1-Ala55Thr
These genetic findings indicated the possibility that the CX3CR1-Ala55Thr variant might associate with the pathogenesis of SCZ and ASD. We therefore preformed 3D structural modeling of CX3CR1 including a membrane, the CX3CR1 ligand fractalkine, and a heterotrimeric G protein (Figure 2a). As seen for other GPCR proteins, the CX3CR1 model has seven transmembrane (TM) helices that pass through the cell membrane, and a small amphipathic α-helix called ‘helix 8’ in the C-terminal cytoplasmic region. Helix 8 is reported to be important for signal transduction and interacts with the heterotrimeric G protein and other diverse signaling molecules.61, 62 Ala55 is located in TM1; however, it interacts with the helix 8 and not with the membrane. Several non-polar residues of TM1 and helix 8 form a hydrophobic core: Leu51, Val52, and Ala55 of TM1, and Phe300, Tyr303, Leu304, and Leu307 of helix 8 (Figure 2b). The variant of Ala to Thr in which residue 55 is changed from a hydrophobic (alanine) to a hydrophilic residue (threonine) may weaken the hydrophobic interaction between TM1 and helix 8, and destabilize the conformation of helix 8 (Figures 2b and c). This conformational change of helix 8 may then disturb the interaction with Gα and downstream signaling pathways.

3D model of the structure of the CX3CR1-Ala55Thr mutant. (a) 3D model of the structure of CX3CR1, fractalkine, a heterotrimeric G protein (Gα, Gβ, Gγ) and the cellular membrane. Three CX3CR1-mutated sites (Ala55, Gly112, Met138) are indicated in dotted black circles. (b) Enlarged view of the 55th residue of wild-type CX3CR1. (c) Enlarged view of the 55th residue of the CX3CR1-Ala55Thr mutant.
Functional analysis of the effect of CX3CR1-Ala55Thr on Akt phosphorylation
We then investigated the effect of CX3CR1-Ala55Thr on CX3CR1 signaling. We first generated expression vectors encoding CX3CR1 WT or the Ala55Thr mutant conjugated with mCherry, and assessed their subcellular localization in HEK293 cells. There were no obvious differences in subcellular localization between CX3CR1 WT and its Ala55Thr mutant (Figure 3a). We then investigated the effects of Ala55Thr mutation on CX3CR1 signaling in HEK293 cells. It is well established that the chemokine fractalkine activates the Akt pathway through CX3CR1.39, 41, 63 HEK293 cells were used for the present study because it has been reported that the level of fractalkine binding with non-transfected HEK293 cells is very low.64 Overexpression of CX3CR1 WT in HEK293 cells increased the phosphorylation level of Akt in the presence of fractalkine. However, treatment with fractalkine did not induce Akt phosphorylation when the cells were transfected with the CX3CR1-Ala55Thr mutant (Figure 3b). These results suggest that CX3CR1-Ala55Thr mutation inhibits fractalkine-CX3CR1 signaling.

Effect of CX3CR1-Ala55Thr on Akt phosphorylation-mediated signaling. (a) Subcellular localization of CX3CR1 wild-type (WT) or its Ala55Thr mutant. mCherry-fused CX3CR1 WT or mutant-expressing vectors were transfected into HEK293 cells and cultured for 36 h before immunofluorescent analysis. DIC, differential interference contrast. Scale bar, 25 μm. (b) Ala55Thr mutation in CX3CR1 inhibits Akt phosphorylation upon fractalkine (FKN) treatment. HEK293 cells transfected with an mCherry-conjugated CX3CR1 WT or Ala55Thr-expressing vector were cultured for 36 h, and then the cells were stimulated with the CX3CR1 ligand fractalkine for 15 min. Cell lysates were analyzed by western blotting using the indicated antibodies. The data are expressed as means±s.e.m. and statistical significance was tested with analysis of variance followed by Tukey–Kramer’s multiple comparison test. **P<0.01, *P<0.05. n=5.
Discussion
Our results of the genetic association of the Ala55Thr variant in CX3CR1 with both SCZ and ASD; the conformational change in the CX3CR1-Ala55Thr mutant compared with CX3CR1 WT that was predicted by in silico 3D structural modeling; and the downregulation of fractalkine-CX3CR1 signaling by the Ala55Thr mutant in vivo support the hypothesis that this variant in CX3CR1 might be a plausible candidate causal variant in SCZ and ASD etiopathologies. Rare variants with a large effect may explain a part of the missing heritability.9 In this regard, we detected one condition-related SNV through targeted resequencing of coding exons in CX3CR1 for 562 Japanese SCZ or ASD patients and subsequent genetic association analysis in a sample comprising 7054 unrelated individuals. A statistically significant association was found between Ala55Thr and SCZ, ASD and both phenotypes (odds ratio=7.2, 13.1 and 8.3, respectively) (Table 2; Supplementary Table S1). Inheritance for cases with Ala55Thr was either from an unaffected father to son or of unknown origin (Supplementary Figure S1), suggesting variable penetrance. This finding is consistent with recent genetic studies; deleterious alleles are likely to be especially rare because of purifying selection.9 Inherited-truncating variants especially in genes that are closely involved in neurodevelopment are highly enriched in patient populations.13, 65, 66, 67, 68 Thus CX3CR1-Ala55Thr could increase susceptibility to the development of a neuropsychiatric disorder.
Modeling of the 3D structure of the CX3CR1 WT and its Ala55Thr mutant suggested that the Ala55Thr mutation might destabilize the hydrophobic interaction between TM1 and helix 8. The canonical mechanism of signal transduction initiated by GPCRs involves the activation of a heterotrimeric G protein by an agonist-occupied receptor.60, 63 In addition to the seven transmembrane helices, conformational dynamics of helix 8 in the cytoplasmic C terminus has been suggested to have an important role in its intracellular signal transduction.61, 62 Functional analysis using in vitro assays indicated that the Ala55Thr variant in CX3CR1 might decrease the fractalkine-CX3CR1 signaling. Its signaling impairment has been reported to influence microglial function.69, 70 On the basis of the complementary expression of fractalkine on neurons and CX3CR1 on microglia, it has been proposed that the neuron signaling to microglia is mediated through the CX3CR1 receptor.31 Fractalkine-CX3CR1 signaling might limit microglia toxicity through induction of PI3K/Akt and ERK1/2 phosphorylation.45, 71, 72, 73 As there are no differences in fractalkine and CX3CR1 expression in brain tissue between patients with and without neurodegenerative diseases,69 it is conceivable that functional change in fractalkine-CX3CR1 signaling could contribute to the pathophysiology of neurodevelopmental disorders by modulating microglial activation.
There are several limitations of this study. First, although the data presented here support the hypothesis that rare variants in CX3CR1 contribute to the neurodevelopmental disorders of SCZ and ASD, it is clear that multiple pathways regulate synapse connectivity in the brain, which account for complex genetic influences on SCZ and ASD pathogenesis. Of note, the Ala55Thr variant in CX3CR1 was present in an extremely small fraction (<0.5%) of the tested patients with neurodevelopmental disorders. The idea of performing a study that focuses on coding variants of a gene in terms of their contribution to a disease has been supported by a recent finding that rare and low-frequency coding variants contribute to the genetic architecture of a complex trait;11 however, the promoter, untranslated regions or intronic regions of CX3CR1 that our variation screening did not cover, also potentially contain disease-associated regions. Second, regarding the genotype–phenotype evaluations, we could neither fully explore the impact of the novel variant nor monitor variant segregation due to limited access to the detailed clinical phenotypes of patients and to lack of DNA from patient family members. Although the father carrying CX3CR1-Ala55Thr had neither history of mental disorders nor symptom causing clinically significant impairments in social, occupational or other important areas of current functioning, possibility that the father might present personality traits, which have been indicated as the ‘Broader Autism Phenotype’ still remains.74 Careful assessments of proband and family members will provide insight into the underlying genetic mechanisms and determine the impact of rare CX3CR1 variants on psychopathology. Finally, we only demonstrated a reduction in the Akt phosphorylation-mediated signaling by CX3CR1-Ala55Thr using HEK293 cells. Additional factors produced by microglia may ameliorate the disturbed function of CX3CR1 in vivo. Further analysis performed in microglia will be needed to assess the exact molecular mechanisms and networks affected by CX3CR1 variants in SCZ and ASD.
In conclusion, individuals with the Ala55Thr variant of CX3CR1 might increase the susceptibility to developing the neurodevelopmental disorders such as SCZ and ASD. To our knowledge, our findings provide the first genetic evidence in a microglia-specific gene for association with neurodevelopmental disorders. A deeper understanding of genetic risk factors and disease pathobiology may lead to major health benefits through the development of methods for the prevention, diagnoses and treatment of such diseases.
References
Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N . Autism spectrum disorders and childhood-onset schizophrenia: clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry 2009; 48: 10–18.
Sullivan PF, Daly MJ, O'Donovan M . Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012; 13: 537–551.
Sullivan PF, Magnusson C, Reichenberg A, Boman M, Dalman C, Davidson M et al. Family history of schizophrenia and bipolar disorder as risk factors for autism. Arch Gen Psychiatry 2012; 69: 1099–1103.
Craddock N, Owen MJ . The Kraepelinian dichotomy—going, going... but still not gone. Br J Psychiatry 2010; 196: 92–95.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–994.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381: 1371–1379.
Kryukov GV, Pennacchio LA, Sunyaev SR . Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet 2007; 80: 727–739.
McClellan J, King MC . Genetic heterogeneity in human disease. Cell 2010; 141: 210–217.
Gibson G . Rare and common variants: twenty arguments. Nat Rev Genet 2011; 13: 135–145.
Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA 2014; 111: E455–E464.
Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR et al. Rare and low-frequency coding variants alter human adult height. Nature 2017; 542: 186–190.
Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, Woolfrey KM . Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 2011; 14: 285–293.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215.
Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry 2014; 19: 872–879.
Hommer RE, Swedo SE . Schizophrenia and autism-related disorders. Schizophr Bull 2015; 41: 313–314.
Bourgeron T . From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 2015; 16: 551–563.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005; 8: 752–758.
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J . Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 2009; 29: 3974–3980.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333: 1456–1458.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012; 74: 691–705.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 2013; 16: 543–551.
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O . Neuroinflammation and psychiatric illness. J Neuroinflammation 2013; 10: 43.
Prinz M, Priller J . Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 2014; 15: 300–312.
Chung WS, Welsh CA, Barres BA, Stevens B . Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 2015; 18: 1539–1545.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530: 177–183.
van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry 2008; 64: 820–822.
Goudriaan A, de Leeuw C, Ripke S, Hultman CM, Sklar P, Sullivan PF et al. Specific glial functions contribute to schizophrenia susceptibility. Schizophr Bull 2014; 40: 925–935.
Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013; 70: 49–58.
Bergon A, Belzeaux R, Comte M, Pelletier F, Herve M, Gardiner EJ et al. CX3CR1 is dysregulated in blood and brain from schizophrenia patients. Schizophr Res 2015; 168: 434–443.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384.
Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA 1998; 95: 10896–10901.
Tremblay ME, Lowery RL, Majewska AK . Microglial interactions with synapses are modulated by visual experience. PLoS Biol 2010; 8: e1000527.
Limatola C, Ransohoff RM . Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front Cell Neurosci 2014; 8: 229.
Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 2014; 17: 400–406.
Sophie F, Laurence M, Dominique C, Céline V, Emmanuelle G, Brigitte A et al. Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science 2000; 287: 2274–2277.
Arli B, Irkec C, Menevse S, Yilmaz A, Alp E . Fractalkine gene receptor polymorphism in patients with multiple sclerosis. Int J Neurosci 2013; 123: 31–37.
Lopez-Lopez A, Gamez J, Syriani E, Morales M, Salvado M, Rodriguez MJ et al. CX3CR1 is a modifying gene of survival and progression in amyotrophic lateral sclerosis. PLoS ONE 2014; 9: e96528.
Tuo J, Smith BC, Bojanowski CM, Meleth AD, Gery I, Csaky KG et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J 2004; 18: 1297–1299.
Moatti D, Faure S, Fumeron F, Amara Mel-W, Seknadji P, McDermott DH et al. Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood 2001; 97: 1925–1928.
Lee S, Abecasis GR, Boehnke M, Lin X . Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 2014; 95: 5–23.
Heilker R, Wolff M, Tautermann CS, Bieler M . G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov Today 2009; 14: 231–240.
Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ . The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J Immunol 2000; 165: 397–403.
Chandrasekar B, Mummidi S, Perla RP, Bysani S, Dulin NO, Feng L et al. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J 2003; 373: 547–558.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 2006; 9: 917–924.
Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS et al. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am J Physiol Heart Circ Physiol 2006; 291: H2836–H2846.
Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 2009; 113: 963–972.
Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun 2015; 6: 8018.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Schwarz JM, Cooper DN, Schuelke M, Seelow D . MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014; 11: 361–362.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014; 42: D980–D985.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009; 84: 524–533.
Purcell S, Cherny SS, Sham PC . Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003; 19: 149–150.
Zheng Y, Qin L, Zacarias NV, de Vries H, Han GW, Gustavsson M et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016; 540: 458–461.
Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS et al. Crystal structure of the beta2 adrenergic receptor–Gs protein complex. Nature 2011; 477: 549–555.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402.
Kawabata T . HOMCOS: an updated server to search and model complex 3D structures. J Struct Funct Genomics 2016; 17: 83–99.
Sali A, Blundell T . Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234: 779–815.
Kawabata T, Nishikawa K . Protein structure comparison using the markov transition model of evolution. Proteins 2000; 41: 108–122.
Wada M, Kanamori E, Nakamura H, Fukunishi Y . Selection of in silico drug screening results for G-protein-coupled receptors by using universal active probes. J Chem Inf Model 2011; 51: 2398–2407.
Burg JS, Ingram JR, Venkatakrishnan A, Jude KM, Dukkipati A, Feinberg EN et al. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 2015; 347: 1113–1117.
Huynh J, Thomas WG, Aguilar M-I, Pattenden LK . Role of helix 8 in G protein-coupled receptors based on structure–function studies on the type 1 angiotensin receptor. Mol Cell Endocrinol 2009; 302: 118–127.
Sato T, Kawasaki T, Mine S, Matsumura H . Functional role of the C-terminal amphipathic helix 8 of olfactory receptors and other G protein-coupled receptors. Int J Mol Sci 2016; 17: 1930.
Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci USA 2010; 107: 13666–13671.
Cybulsky MI, Hegele RA . The fractalkine receptor CX3CR1 is a key mediator of atherogenesis. J Clin Invest 2003; 111: 1118–1120.
Mowry BJ, Gratten J . The emerging spectrum of allelic variation in schizophrenia: current evidence and strategies for the identification and functional characterization of common and rare variants. Mol Psychiatry 2013; 18: 38–52.
Hoischen A, Krumm N, Eichler EE . Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci 2014; 17: 764–772.
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014; 506: 185–190.
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K et al. Excess of rare, inherited truncating mutations in autism. Nat Genet 2015; 47: 582–588.
Hulshof S, van Haastert E, Kuipers H, van den Elsen P, De Groot C, van der Valk P et al. CX3CL1 and CX3CR1 expression in human brain tissue: noninflammatory control versus multiple sclerosis. J Neuropathol Exp Neurol 2003; 62: 899–907.
Wolf Y, Yona S, Kim K-W, Jung S . Microglia, seen from the CX3CR1 angle. Front Cell Neurosci 2013; 7: 26.
Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB . Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J Immunol 1999; 163: 1628–1635.
Shuzhen C, Defang L, Wolfgang JS, Jeffrey KH . TGF-β1 upregulates CX3CR1 expression and inhibits fractalkine-stimulated signaling in rat microglia. J Neuroimmunol 2002; 133: 46–55.
Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 2011; 286: 32713–32722.
Billeci L, Calderoni S, Conti E, Gesi C, Carmassi C, Dell’Osso L et al. The broad autism (Endo)phenotype: neurostructural and neurofunctional correlates in parents of individuals with autism spectrum disorders. Front Neurosci 2016; 10: 346.
Acknowledgements
We are grateful to all the patients, their families, and control individuals who contributed to this study. This work was supported by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Ministry of Health, Labour, and Welfare of Japan; the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from Japan Agency for Medical Research and Development (AMED); the Strategic Research Program for Brain Sciences of AMED; the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) project of AMED; Grant-in-Aid for Scientific Research on Innovative Areas ‘Glial assembly: a new regulatory machinery of brain function and disorders’ and Innovative Areas ‘Comprehensive Brain Science Network’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ishizuka, K., Fujita, Y., Kawabata, T. et al. Rare genetic variants in CX3CR1 and their contribution to the increased risk of schizophrenia and autism spectrum disorders. Transl Psychiatry 7, e1184 (2017). https://doi.org/10.1038/tp.2017.173
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.173
This article is cited by
-
The genetic architecture of schizophrenia: review of large-scale genetic studies
Journal of Human Genetics (2023)
-
Microglial contribution to the pathology of neurodevelopmental disorders in humans
Acta Neuropathologica (2023)
-
Cerebral dysfunctions caused by sepsis during ageing
Nature Reviews Immunology (2022)
-
In vivo imaging translocator protein (TSPO) in autism spectrum disorder
Neuropsychopharmacology (2022)
-
Linking Inflammation, Aberrant Glutamate-Dopamine Interaction, and Post-synaptic Changes: Translational Relevance for Schizophrenia and Antipsychotic Treatment: a Systematic Review
Molecular Neurobiology (2022)
