
Abstract
Panic disorder (PD), a complex anxiety disorder characterized by recurrent panic attacks, represents a poorly understood psychiatric condition which is associated with significant morbidity and an increased risk of suicide attempts and completed suicide. Recently however, neuroimaging and panic provocation challenge studies have provided insights into the pathoetiology of panic phenomena and have begun to elucidate potential neural mechanisms that may underlie panic attacks. In this regard, accumulating evidence suggests that acidosis may be a contributing factor in induction of panic. Challenge studies in patients with PD reveal that panic attacks may be reliably provoked by agents that lead to acid–base dysbalance such as CO2 inhalation and sodium lactate infusion. Chemosensory mechanisms that translate pH into panic-relevant fear, autonomic, and respiratory responses are therefore of high relevance to the understanding of panic pathophysiology. Herein, we provide a current update on clinical and preclinical studies supporting how acid–base imbalance and diverse chemosensory mechanisms may be associated with PD and discuss future implications of these findings.
Similar content being viewed by others

Introduction
Panic disorder (PD) is characterized by spontaneous and recurrent panic attacks that consist of incapacitating periods of acute-onset respiratory, cardiovascular, gastrointestinal, autonomic and cognitive symptoms. PD—which occurs in 6% of Americans1—typically begins in the second decade of life2 and exhibits a peak prevalence in the third and fourth decades of life.3 Thus, this condition is second only to major depressive disorder in terms of associated debility among psychiatric conditions in the United States.4 Importantly, PD also represents an independent risk factor for suicidality in diagnostically and demographically heterogeneous clinical populations5 and increases the risk of developing other anxiety disorders and secondary mood disorders.2 Yet, many patients suffering from PD are not clinically identified and frequently, do not receive even minimally effective treatment.6 Even still, available psychopharmacologic treatments (for example, selective serotonin reuptake inhibitors, benzodiazepines) and psychotherapies (for example, cognitive behavioral therapy, prolonged exposure therapy, psychodynamic psychotherapy) or the combination of psychotherapy+pharmacotherapy are often only modestly efficacious (for example, Cohen’s d=0.4–0.6)7, 8 and in some cases (for example, benzodiazepines) may be associated with treatment-specific side effects or risks such as sedation or the risk of dependence or tolerance.
Studies elucidating the pathoetiologic mechanisms of PD are urgently needed to reduce morbidity and mortality. Despite the prevalence, as well as associated morbidity and mortality of PD, relatively little regarding the neuropathophysiology of this condition is known. PD is highly heterogenous with variable symptom profile and intensity in panic episodes experienced by the same individual and across patients. According to the DSM-5,9 recurrent panic attacks in PD are categorized as being either spontaneous (unexpected) or cued (expected). Collective evidence from challenge studies in the laboratory, neuroimaging, symptomology, treatment responses and translational animal models have led to an increased understanding of PD.10, 11, 12, 13, 14 Accumulating evidence suggests that expected panic attacks are triggered by exteroceptive threats (that is, a panic attack context or other unrelated stressors) while spontaneous panic attacks may be provoked by interoceptive sensory triggers caused by fluctuations in the internal homeostatic milieu. An important internal homeostatic trigger for the genesis of panic attacks, supported by an emerging body of work, is acid–base imbalance and associated pH chemosensory mechanisms. Largely founded on panic provocation studies with agents promoting homeostatic pH imbalance and related to the false suffocation alarm theory, the role of acid–base and chemosensory systems in panic provides strong scientific insights on the genesis of uncued panic attacks which may sensitize fear-arousal-stress regulatory circuits to other triggers leading to full-blown PD (Figure 1, cycle of panic). Given the high relevance of interoceptive mechanisms in PD, this review provides an update on our current knowledge and understanding of the role of pH imbalance and chemosensory targets in PD. Although excellent reviews on this topic have appeared previously,15, 16, 17 here we focus on (1) current status on pH homeostasis, clinical studies of acid–base physiology and pathophysiology in patients with PD (2) preclinical rodent models of PD, especially those focusing on interoceptive pH imbalance and acid-chemosensory systems recruited in panic-like behaviors, and last (3) synthesize these findings to develop a working neurobiological model of PD that involves dysregulation of central acid sensing and associated circuitry, and finally, (4) translational relevance of these data, gaps in understanding and future implications with a discussion on neuropharmacologic interventions in patients with PD.

Potential pathogenesis of uncued and cued panic attacks in panic disorder: Initial unexpected attacks may result from an acid/base imbalance or from altered chemosensory mechanisms that represent a ‘threat to homeostasis’. Although the exact origin of pH disturbance is unknown, it may arise due to genetic predisposition, respiratory abnormalities and other factors. This may produce a state of alarm and subsequent activation of threat response systems leading to elevated fear, cardiovascular and respiratory symptoms which, phenomenologically, constitute a panic attack. Further, experiencing uncued panic attacks may sensitize threat response systems to exteroceptive triggers such as stress, panic context and associated phobic cues leading to cued panic attacks. Persistence of uncued and cued panic attacks results in full-blown panic disorder.
Acidosis, an interoceptive trigger in panic: evidence for relationship between pH disturbances in PD
Many clinical and preclinical studies have focused on dysregulation of central fear circuitry that includes components of the limbic network, which involves connections between the amygdala and the anterior cingulate cortex (Broadman’s area 25, 24/32) as well as midbrain regions including the periaqueductal gray matter in panic.10, 14 These studies have provided useful information relevant to cued panic attacks and phobias in PD subjects; however, the genesis of unexpected panic attacks still remains elusive to panic researchers. James18 first proposed that feelings and emotion can derive from interoceptive sensing of our body states. These internal triggers and interoceptive chemosensory pathways are of particular relevance to PD as initial attacks come ‘out of no-where’. Moreover, accumulating evidence supports a principal role of pH homeostasis in panic physiology and suggests that acidosis may be an interoceptive trigger for panic attacks. Consistent with this, a recent study with ambulatory monitoring, a valid approach for studying spontaneous panic, reported pH disturbances and altered respiratory rhythms in subjects during the final minutes before the onset of a panic attack.19 Well-characterized and clinically relevant methods of inducing panic, such as CO2 inhalation or sodium lactate infusion, cause acid–base disturbances.16 Importantly, CO2 inhalation and sodium lactate infusion stimulate respiration, which is itself tightly regulated by pH.20 Thus, it is intriguing that despite their disparate effects, respiratory acidosis and metabolic alkalosis, both CO2 inhalation and sodium lactate administration lead to extracellular and intracellular acidosis in the brain.16, 21 In addition, neuroimaging studies also raise the possibility of dysregulated acid–base buffering and increased plasma and brain lactate responses to metabolic challenges in PD.22, 23, 24 There is also a high prevalence of hyperventilation and other respiratory abnormalities among patients with PD.21, 25, 26 The link between panic attacks and pH disturbances also forms the basis of the false suffocation alarm theory of spontaneous panic, where CO2 hypersensitivity may exist due to a malfunctioning suffocation alarm monitor.27 Below we discuss specific areas of investigation that support the panic-pH link.
Brain pH in patients with PD: evidence from neuroimaging
Neuroimaging studies on PD patients support a role of homeostatic pH disturbances in panic physiology (Table 1). The majority of these studies have focused on lactate responses to homeostatic or activity-dependent challenges. Given the close relationship between lactate and pH in the brain, the findings are consistent with a model of brain metabolic and pH dysregulation associated with altered function of acid-sensitive fear circuits as a trait vulnerability factor in PD. Exaggerated activity-dependent brain lactate responses are observed in PD patients, even remitted patients, as compared with healthy controls, suggestive of underlying pH abnormalities.23, 28 Further, activity-dependent changes in glutamate–glutamine were smaller in PD patients.28 Others have shown increased (and more prolonged) brain lactate levels compared with healthy comparison subjects during sodium lactate infusion, although resolution was limited.29 In a subsequent study, the greater and prolonged brain lactate rises in the insula in patients with PD during and following lactate infusion30 were observed—a finding of great importance given the central role of the insula in interoceptive pathways.31 Interestingly, differential brain metabolic responses fail to normalize following treatment with the selective serotonin reuptake inhibitor fluoxetine suggesting that abnormal metabolic lactate responses represent a trait feature of PD. Accentuated increases in brain lactate levels have also been reported in PD subjects during hyperventilation, that promotes pH imbalance, as compared with comparison subjects,22 suggesting that increased lactate possibly reflects altered cerebral blood flow in response to hypocapnia evoked by increased breathing. Using phosphorous (31P) spectroscopy, pH dynamics during hyperventilation were assessed by Friedman et al.,32 and reported an abnormal pH-buffering capacity in PD. Very recently, CO2-evoked activation in the brain stem area was reported in panic patients, controls and divers (representing a group with reduced CO2 sensitivity).33 The authors reported significant increases in activation within brain stem and insular areas in panic patients as compared with controls and diver groups supporting the role of these areas in interoceptive sensing and processing in PD. Recently, new magnetic resonance imaging techniques such as the T1 relaxation in the rotating frame (T1ρ) have been developed and have facilitated greater pH sensitivity and improved temporal and spatial resolution over 31P spectroscopy.34 Increased T1ρ was observed in PD patients in the occipital cortex as compared with healthy controls35 consistent with abnormal pH regulation in PD. Interestingly, the magnitude of the T1ρ response correlates with the severity of anxiety symptoms in patients with PD (but not in healthy subjects). Thus, it is increasingly apparent that this new imaging technique will allow for greater understanding of pH-dependent changes in PD. Finally, although studies of panic symptom provocation are still needed, evidence from functional magnetic resonance imaging and functional magnetic resonance imaging supports differences in metabolic activity or other factors such as cerebral blood flow may lead to altered pH homeostasis in PD.
Symptom provocation challenge studies with agents producing acid–base imbalance
Panic attacks can be induced in PD individuals by a variety of agents such as CO2,36, 37 sodium lactate,38 doxapram,39 isoproterenol,40 caffeine,41 yohimbine,42 cholecystokinin (CCK-4 and agonists),43 the benzodiazepine receptor antagonist, flumazenil,44 serotonin receptor agonists45 and opioid receptor antagonists.46 It is important to note, however, that homeostatic perturbations such as CO2, lactate and doxapram that cause direct or compensatory pH shifts reliably evoke panic attacks that closely resemble spontaneous panic attacks. Furthermore, as opposed to other challenges, these agents induce panic attacks specifically in PD subjects versus other disorders such as depression, premenstrual dysphoric disorder, generalized anxiety disorder and posttraumatic stress disorder.47, 48 Panic provocation agents linked to homeostatic pH imbalance are discussed below.
CO2 inhalation
Inhalation of CO2—a commonly studied interoceptive stimulus—produces intense fear, autonomic and respiratory responses that can evoke panic attacks in individuals with PD. For this reason, CO2 is frequently used as a biological challenge and pathological marker of PD.11, 36 First described in 1951 by Cohen and White,37 CO2 inhalation is established as a reliable panicogen in patients with PD.11, 36, 49, 50
The partial pressure of CO2 in the blood increases following CO2 inhalation challenge. In addition, and of direct relevance to central nervous system (CNS) physiology, CO2 readily crosses the blood–brain barrier and is sensed by H+ and CO2 chemoreceptors in the CNS and periphery.51 In the extracellular fluid, CO2 is hydrolyzed to carbonic acid (H2CO3) by carbonic anhydrase which readily dissociates into bicarbonate (HCO3-) and H+.51 The resulting acidosis is thought to be the trigger for the panic symptoms caused by this challenge including hyperventilation and increased blood pressure.52 Klein puts forth in his false suffocation theory that hyperventilation may have a protective role to combat attacks caused by increases in CO2 (acidosis).27 However, this is a faulty response because the respiratory alkalosis caused by hyperventilation is always associated with a compensatory metabolic acidosis produced by tissue buffering systems that release H+ ions.21 Panic challenge studies with acetazolamide shed light on the role of protons as effector molecules for generating panic responses; acetazolamide, a carbonic anhydrase inhibitor, blocks the facilitated conversion of CO2 to bicarbonate and H+, leading to increases in CO2 concentrations. Interestingly, administration of intravenous acetazolamide fails to induce panic attacks in patients with PD53, 54 suggesting that H+ ions, rather than CO2 per se may facilitate panicogenesis.
Currently, two CO2 inhalation techniques are used in panic challenge studies. In the first, steady-state inhalation, a low concentration of CO2 (5–7.5%) is inhaled for approximately 1–20 min or until a panic attack occurs. In the second approach, individuals inhale a high concentration of CO2 (35%).36 The advantage of modeling CO2-induced panic is that these CO2-induced panic attacks closely resemble spontaneous panic attacks and the attacks resolve quickly.11 Interestingly, although PD is twice as likely to occur in women,3 sex differences in CO2-reactivity are less clear. Although there is some evidence that women report greater fear and anxiety following a CO2 challenge,55, 56, 57 not all studies have observed gender effects.50, 58, 59
CO2 inhalation has also been useful for exposure-based treatments in patients with PD60, 61 and has been utilized for validation of current treatments such as selective serotonin reuptake inhibitors: paroxetine, sertraline, fluvoxamine62 and benzodiazapine alprazolam.63 In addition, screening of potential anti-panic medications such as CRF1 receptor antagonist, R317573,64 GABA agonist: zolpidem63 and neurokinin-1 receptor antagonist: vestipitant65 has also been conducted using this challenge. Thus, CO2 inhalation appears to have utility for testing the efficacy of pharmacotherapeutic agents and for identifying vulnerability to PD.
Sodium lactate infusion
In addition to CO2, sodium lactate is a reliable panicogen38 frequently used in challenge paradigms. A masked intraveneous infusion of a 0.5 M sodium lactate (10 ml kg−1) produces panic attacks in vulnerable individuals.38, 66 Lactate-induced panic attacks, like CO2-induced panic attacks, phenomenologically mirror spontaneous panic attacks (that is, symptoms of dyspnea, generalized fear, a desire to flee and fear of losing control.67 Clinically, susceptibility to lactate-induced panic attacks are frequently used as treatment outcome measures for psychopharmacologic treatments.68, 69, 70
A byproduct of cellular metabolism, lactate serves as an energy source for neurons,71 and alters systemic acid–base balance. Pertinent to lactate infusions, lactate can cross the blood–brain barrier through monocarboxylate transporters and there is evidence that lactate becomes a significant fuel source in the brain when elevated in blood.72 When administered intravenously to lower primates, lactate decreases brain pH73 as H+ is co-transported with lactate via monocarboxylate transporters. Although lactate infusion may evoke acidosis, a direct role of pH in lactate-evoked panic has not been demonstrated. Interestingly, patients with PD show exaggerated lactic acid production in response to alkalosis evoked by sodium lactate infusion suggestive of increased compensatory drive and impaired acid–base buffering in these individuals.17 Other studies reported that a rapid overload of sodium and resultant acute hypernatremia may contribute to sodium lactate-evoked panic since hypertonic saline (3%) facilitated panic symptoms similar to 0.5 M sodium lactate.74 An interesting observation in the study was the induction of mild acidosis by hypertonic saline while sodium lactate-evoked hyperventilation and associated alkalosis, although specific parameters such as blood pCO2 were not measured. Lactate-evoked panic attacks do not recruit neuroendocrine responses as a dissociation between autonomic activation and cortisol has been reported in ‘panickers’ following sodium lactate.75 Potential downstream mechanisms for lactate sensitivity in PD are not clear. Involvement of GABAergic system has been suggested by effective blockade of lactate-evoked panic in subjects treated with gabapentin,76 while presynaptic, α2adrenergic agonist, clonidine had partial effects. Additionally, concentrations of endogenous neuroactive steroid modulators of the GABAA receptor, allopregnanolone and pregnanolone are decreased in patients with PD during lactate-evoked panic.77 Elegant preclinical studies by Shekhar and colleagues have highlighted the role of circumventricular organs (CVOs), hypothalamic GABA, angiotensin and orexin systems in sodium lactate-evoked panic responses (see section ‘Sodium lactate rodent model and hypothalamic GABAergic and acid-chemosensitive orexin targets’). Thus, the underlying mechanism(s) or a direct role of acidosis in sodium lactate-induced panic attacks has not been elucidated to date, and while lactate may contribute to decreased brain pH, the exact effector in lactate-evoked panic in PD subjects is still unclear.
Doxapram
Doxapram, a respiratory stimulant first synthesized in 1962 (Ward and Franko78) has been examined in the management of acute respiratory failure during the 1960s and 1970s and likely had a specific role in the treatment of individuals with chronic obstructive pulmonary disease. Specifically, administration of doxapram increases tidal volume and ventilation frequency.79 Clinically, doxapram use is primarily limited to post-anesthesia shivering prophylaxis and stimulation of respiratory drive in premature infants. However, its use is also associated with second-degree atrioventricular block and QTc prolongation. Regarding patients with PD, doxapram has been examined in several studies, given its tendency to cause hyperventilation. In this challenge, doxapram (0.5 mg kg−1, IV)39, 80, 81 produces panic attacks that phenomenologically mirror spontaneous panic attacks with associated hyperpnea, tachycardia, increased blood pressure and fear symptoms. Contribution of forebrain regions in doxapram-evoked panic is supported by recent studies where doxapram was administered to PD patients and healthy subjects before positron emission tomography with 18F-deoxyglucose.82 Cardiac responses were accentuated and patients with PD exhibited decreased prefrontal activity (relative to controls) and increased activity within the cingulate gyrus and amygdala, suggesting a failure to activate prefrontal inhibitory structures in patients with PD.
The underlying mechanism, specifically a role of pH in doxapram-evoked panic has not been established. Hyperventilation induces alkalosis, which has been reported to evoke a compensatory increase in lactic acid release; a response that is exaggerated in PD patients.17 It is unclear, however, whether this compensatory increase in acidosis is associated with doxapram-evoked panic attacks. There is evidence that the effects of doxapram may be related to the inhibition of TASK-1 and TASK-3 acid-sensitive potassium channels located in brain stem serotonergic neurons.83, 84 In this regard, inhibition of the TASK-1 and TASK-3 channels could increase the excitability of brain stem pH-sensitive neurons and may link the panicogenic action of CO2 inhalation and doxapram administration.85 In addition, increased respiratory drive by doxapram may aggravate a pre-existing respiratory abnormality in PD. As for lactate, a potential role of pH and acidosis in panic provocation by doxapram may be speculated, however, direct evidence for this link is currently lacking.
Genetics
A strong contribution of genetics and family history in PD prevalence was first reported by Crowe and colleagues.86 In support of a genetic component in vulnerability to interoceptive triggers and PD, higher sensitivity to 35% CO2 was observed in first-degree relatives of patients with PD.87 CO2 hypersensitivity has been proposed as a genetic risk and disease-specific trait marker for PD88 also supported by twin studies.89, 90 Importantly, a distinction between genetic vulnerability to CO2 hypersensitivity versus trait anxiety experienced pre-CO2 inhalation was found suggesting that there are specific genetic factors associated with responsivity to stimulation via CO2 versus factors related to underlying trait anxiety.91
However, as PD does not develop in all individuals with CO2 hypersensitivity, it underscores the relevance of other risk factors for development of PD. A combination of genetic factors and early adversity such as childhood parental loss may determine hypersensitivity to CO2 and PD.92 An interesting preclinical study in cross-fostered mice pups revealed persistent expression of enhanced CO2-evoked respiratory responses in mice with a history of interference with dam–pup interactions suggestive of significant gene-by-environment effects on heightened CO2 sensitivity.93 In any case, hypersensitivity to elevated CO2 may help identify childhood groups at familial risk for subsequent development of PD94, 95 Interestingly, association of polymorphisms within the tryptophan hydroxylase-2 (TPH-2) gene and CO2 responses is observed suggestive of a role of the serotonergic (5-HT) system in the effects of CO2.96 Accumulating evidence strongly supports an association of polymorphisms in multiple markers of the 5-HT system, including polymorphisms in the gene locus and 3’ polyadenylation site of the serotonin transporter (5-HTT) with PD.97, 98 An association of 5-HT biosynthetic enzyme TPH-2 and 5-HT receptor subtypes R1 and R2 with PD has also been reported.99, 100, 101 The 5-HT system is of interest given its role in the regulation of panic-like behaviors.102 Importantly, evidence of chemosensory serotonergic neurons in the medullary raphe (see section ‘Acid chemosensory serotonergic neurons in the medullary raphe nucleus’) underscores the role of the 5-HT system in translation of pH fluctuations to panic-relevant ventilatory responses.
Lactate sensitivity on the other hand, did not show familial vulnerability.103 However, an association of polymorphism within the exon of the lactate dehydrogenase A gene was reported with CO2 sensitivity where the LDH polymorphism was a risk factor for increased CO2 responses.104 This is relevant, given the role LDH in lactate metabolism and its dependence on cell pH.
A recent study identified two single nucleotide polymorphisms within the acid sensing ion channel 1 (ASIC1) gene, ACCN2 in individuals with PD, which was associated with increased amygdala volume and hyperactivity in these subjects.105 This observation strongly supports an association of altered pH sensing within the amygdala with increased risk for PD. Collectively, all evidence support a strong genetic vulnerability component to interoceptive threats (represented by CO2 inhalation) and polymorphisms in chemosensory targets such as ASICs. However, other environmental factors may be required for the development of symptomology of PD.
Collectively, evidence from neuroimaging, challenge studies for panic provocation and genetics support that pH homeostasis and acid–base disturbances may contribute to PD at least in a large subset of patients with PD. In the sections below, we discuss pH chemosensory mechanisms and our understanding so far on their potential contributions to panic pathophysiology using preclinical models.
pH chemosensory molecules and their relevance to panic pathophysiology: evidence from preclinical studies
Acid chemosensory mechanisms become relevant since they can detect and translate interoceptive pH imbalance that may exist in PD (see above). Given the intense respiratory symptoms experienced during a panic attack, it is logical to conclude that these sensing mechanisms are recruited in panic, since respiration is tightly regulated by pH. Multiple chemosensitive areas within the caudal brain stem such as the retrotrapezoid nucleus, solitary nucleus, locus coeruleus and the medullary raphe nuclei elicit neuronal responses to hypercapnia and regulate respiration.51, 106 Although chemosensory mechanisms for pH are generally regarded as a brain stem phenomenon, studies in the past decade have revealed several non-brain stem regions to participate in pH chemosensation and associated behavioral and physiological responses. Primary among these are the amygdala, dorsomedial/perifornical hypothalamus and the periaqueductal gray, although other structures such as CVOs may also participate as chemosensitive sites.107, 108 In fact, the concerted activity as well as, anatomical and functional links between rostral forebrain and brain stem structures is pertinent to panic attacks, where intense respiratory and psychological symptoms coexist. Figure 2 shows the localization of PD-relevant pH chemosensory targets, based on evidence derived from preclinical studies, and associated circuits that might contribute to panic responses. It is important to note that these regions closely overlap with circuits regulating emotional/behavioral responses, autonomic function and respiration. In addition to regulation of fear and anxiety, these areas send projections to caudal brain stem areas thereby impacting respiratory outcomes. The following section describes specific chemosensory targets as well as panic-relevant preclinical paradigms that support their contributions to PD. Although no animal model can simulate all aspects of panic pathophysiology, the unique feature of panic provocation with acid–base modulators has enabled simulation of human panic in preclinical setting.

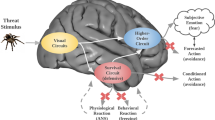
Localization of chemosensory targets and regional circuits contributing to genesis and expression of panic. 1: acid sensing ion channels (ASICs) in the amygdala, 2: orexin neurons in the hypothalamus, 3: serotonergic neurons in the medullary raphe, 4: T-cell death-associated gene-8 receptor in the subfornical organ (SFO), 5: hypoxia-sensitive chemosensory neurons in the periaqueductal gray (PAG). Regions such as the SFO and medullary raphe can directly detect pH fluctuations in the internal milieu, while the hypothalamus, amygdala and PAG in addition to their chemosensory potential also represent key nodes in the processing of external threats, and sensory stimuli. Uncued panic may arise due to homeostatic imbalance in pH in the brain and internal milieu. Acidosis ‘sensed’ by chemosensory mechanisms may be translated to autonomic, behavioral and respiratory symptoms of a panic attack. The amygdala, PAG and the hypothalamus can regulate behavioral and autonomic symptoms of panic, whereas respiratory symptoms may be regulated by brain stem regions such as the medullary raphe and the parabrachial nucleus (PBN) via inputs from the hypothalamus and indirectly from the SFO through the organum vasculosum of the lamina terminalis (OVLT). Many of these structures via thalamic nuclei connect with the insula, a region relevant for interoceptive sensing and shown to be dysfunctional in PD. Cued panic attacks may be an outcome of sensory stimuli and phobic cues associated with previous attacks or stressors relayed via sensory cortices and thalamic nuclei to the amygdala and the hypothalamus. It is important to note the overlap and connectivity between pH chemosensory regions and exteroceptive threat processing areas suggesting that uncued and cued panic may recruit similar underlying circuitry depending on modality of the trigger leading to panic.
Acid-sensing ion channel
The most well-characterized acid-chemosensory target in terms of PD relevance is the acid-sensing ion channel 1 (ASIC-1a), a voltage-insensitive H+-gated cation channel located on neurons in the CNS (reviewed in Wemmie15). ASIC-1a has high levels of expression in the amygdala, dentate gyrus of the hippocampus, cortex, striatum and nucleus accumbens,109, 110 regions that are components of the limbic–corticostriatal loop, which is thought to be involved in assigning emotional valance to external stimuli.109 The contribution of ASIC-1a localized in the amygdala has been studied using translational as well as clinical studies using CO2 inhalation. Mice lacking ASIC-1a show decreased fear responses to CO2 inhalation.111 As shown in that study, CO2 inhalation reduced amygdalar pH to an extent that activates ASIC-1a.111 Furthermore, only control mice, not ASIC-1a knockout mice, freeze in response to lowered amygdala pH (secondary to injection of acidified artificial cerebrospinal fluid). In addition, restoration of ASIC-1a expression in the amygdala of knockout mice is associated with a return of freezing responses to CO2. Collectively, these results suggest that the amygdala is an acid-chemosensitive site and that ASIC-1a within the amygdala mediate CO2-evoked fear responses. However, ASIC-1a knockout mice also demonstrate attenuated fear responses during context and cued fear conditioning109 and exposure to predator odor challenge,112 suggesting that that the regulation of fear by ASIC-1a extends to exteroceptive stimuli and raising the possibility that ASIC-1a may not be selective to interoceptive threats. Genetic studies in humans have linked ASIC polymorphisms to amygdala volume and activity105 suggesting contributions of this chemosensor to amygdala function. In addition, recent studies in humans with bilateral amygdala damage resulting from a rare autosomal recessive disorder, Urbach–Wiethe disease in which there is bilateral destruction of the amygdala, have questioned the necessity of the amygdala in panic responses to interoceptive threats such as CO2.113 As reported in their study, patients with neurodegenerative damage in the amygdala exhibited panic responses following CO2 inhalation challenge. Further, physiological responses to CO2 inhalation such as, ventilation and heart rate, were increased as compared with non-panicking controls and similar to the PD patients. In previous studies, fear responses to exteroceptive threats, such as visiting a haunted house and watching video clips of scary movies were blunted in these patients.114 All together, their results suggest that acid-chemosensory mechanisms in other brain regions may orchestrate fear and panic responses to interoceptive stimuli.
Sodium lactate rodent model and hypothalamic GABAergic and acid-chemosensitive orexin targets
The rodent sodium lactate model of panic pioneered by Shekhar and colleagues provides an excellent translational paradigm in terms of face, predictive and construct validity for sodium lactate challenge in humans (for review see Johnson and Shekhar115). Interestingly, infusing lactate alone does not elicit panic-like behavior in rodents, rather, chronic inhibition of GABAergic tone in the dorsal medial-perifornical hypothalamus is necessary for the expression of lactate-evoked responses. In this regard, rats with tonic inhibition of hypothalamic GABA exhibit intense behavioral, respiratory and cardiovascular responses following sodium lactate infusion.115, 116 Increased anxiety-like behaviors in the social interaction test and elevated-plus maze tests, elevated blood pressure, tachycardia and hyperventilation was observed in GABA-compromised, lactate-infused rats and these behavioral and physiological responses mirror those observed in PD patients challenged with sodium lactate.38 Electrical stimulation of the ventromedial hypothalamus in humans leads to tachycardia and panic (self-reported).117 Along with simulation of panic symptoms, this model is responsive to anti-panic and anxiolytic medications, including the benzodiazepine alprazolam. This model also highlights an important role of sensory CVOs as upstream sites for detection of interoceptive stimuli such as sodium lactate.118 Further investigation revealed that sodium (but not lactate or osmolality) is the primary trigger that is sensed via sodium ion channels in the anterior third ventricle region. Involvement of hypothalamic angiotensin II and orexin in orchestrating panic-like responses to lactate was confirmed using selective antagonists and RNAi probes.119, 120 Orexin expressing neurons in the hypothalamus are of particular interest to panic physiology. In addition to being essential to the effects of sodium lactate, these have been identified as chemosensitive.121, 122 Blunted respiratory responses to hypercapnia were seen in mice lacking prepro-orexin.123 Antagonism of the ORX1 receptor with SB334867 attenuates CO2-induced respiratory responses in mice,123 whereas anxiety and hypertensive responses to CO2 inhalation require activation of the Orexin-1 receptor.124
Thus, converging evidence from the lactate and CO2 inhalation model suggests that orexin antagonists may be an attractive therapeutic option for PD, although prolonged suppression of the orexin system may have its own complications.125
Acid chemosensory serotonergic neurons in the medullary raphe nucleus
The serotonergic raphe neurons in the brain stem detect decreases in pH due to hypercapnia126, 127 and are strategically located near large arteries where they are able to sense levels of CO2 in the blood and quickly initiate behavioral and autonomic responses to maintain homeostasis.128 These serotonergic neurons may be involved in the panic-like responses of CO2 due to their projections to the anterior limbic and prefrontal fear-processing circuits.126 In addition, silencing these pH-sensitive serotonergic neurons in lower animals disrupts chemosensitive responses to CO2 inhalation that impair respiration.129 Recent studies using un-anesthetized in situ perfused decerebrate brain stem preparations suggest that the medullary raphe also contains non-serotonergic, CO2-chemosensitive neurons that are positive for neurokinin-1 receptor, suggesting other targets for CO2-evoked ventilatory responses.130 An interesting study reported significantly lower distress and breathlessness to CO2 inhalation in subjects with acute tryptophan depletion suggesting role of serotonin in promoting aversive respiratory sensations to hypercapnia.131
Taken together, these data suggest a potential relevance for brain stem acid-chemosensory neurons in respiratory distress, of relevance to PD. However, additional studies especially panic-relevant models need to be explored.
Acid-sensing T-cell death-associated gene-8 receptor
The T-cell death-associated gene-8 (TDAG8) receptor, an acid-sensing G-protein-coupled receptor (GPCR) located on immune cells in the CNS and periphery,132, 133, 134 was originally identified by its increased mRNA expression during programmed cell death of mouse thymocytes mediated by T-cell receptor engagement,132 but is increasingly recognized for its putative role in the pathoetiology of panic-like behaviors in lower animals. TDAG8 is a member of the ‘G2A’ group of GPCRs, which includes the G2 accumulation receptor (G2A), ovarian cancer G-protein receptor 1 (OGR-1), and G-protein-coupled receptor 4 (GPR4).132 Although these receptors were originally characterized as lysolipid receptors, they were later found to sense extracellular protons resulting in the stimulation of intracellular signaling pathways.134, 135, 136, 137, 138 Accumulation of cyclic adenosine 5’-monophosphate (cAMP) has been observed in cells transfected with mouse and human TDAG8 cDNA to low extracellular pH.138 To date, TDAG8 is the only proton-sensing GPCR expressed in brain tissue. Recent studies by our group has characterized TDAG8 expression in the CNS, and found it to be enriched in sensory CVOs, which include the subfornical organ, organ vasculosum of the lamina terminalis and the area postrema.108 The sensory CVOs are specialized chemosensory regions that are highly vascularized and lack the blood–brain barrier.139 The sensory CVOs contain cellular contacts with the blood and the cerebrospinal fluid allowing them to relay signals from blood and cerebrospinal fluid to autonomic control centers of the brain.140 Sensory CVOs have been linked to panic via their ability to sense panic-stimuli in the circulation and activate downstream targets via their efferent and afferent projections to prime forebrain and hindbrain.118
Our group has demonstrated that TDAG8 is maximally activated by extracellular pH of 6.5 leading to intracellular increases in cAMP and pCREB in vitro.134 Presence of an acid sensor within brain areas specialized for sensing the internal milieu is important given the relevance of interoceptive sensing in PD. Ongoing studies by our group are investigating the contributions of TDAG8 to panic-relevant responses using TDAG8-deficient mice and translational rodent models of panic phenomenon.108, 141 Preliminary evidence supports attenuation of CO2-evoked fear responses in TDAG8−/− mice.108, 141
Chemosensory neurons in the periaqueductal gray
The recent development of rodent models of panic-like behaviors and physiology highlight the importance of the periaqueductal gray (PAG) in panic responses.14 Electrical and chemical stimulation of the dorsal PAG evokes panic-like responses including freezing, flight, tachycardia, tachypnea and hyperventilation10, 142, 143 in lower animals and stimulation of this region in neurosurgical results in similar behaviors in humans.144 Several structural neuroimaging studies reveal increased gray matter volumes in the midbrain and rostral pons as well as PAG, in PD patients compared with healthy controls.145, 146 Thus, the PAG may represent an attractive site for chemosensory pH sensing and its translation to panic expression. Focal lesions of the PAG do not alter ventilation during normocapnia, however, lead to reduced ventilatory responses to 7% CO2.107 There is now strong evidence that the dorsal PAG contains chemosensitive neurons that may be intrinsically sensitive to O2 reduction, a hypoxia-sensitive alarm. Intravenous potassium cyanide, which produces anoxia, produced panic-like responses such as freezing and flight147 and when paired with 8 or 13% CO2 inhalation, enhances evoked flight responses and, independently facilitates the panic-like responses of rats during electrical PAG stimulation. In addition, electrical lesioning of the PAG attenuates, if not completely abolishes, panic-like responses, raising the possibility that the PAG harbors an anoxia-sensitive suffocation alarm in which dysfunction causes hypersensitivity to CO2. Given the extensive clinical and preclinical studies supporting a role of the PAG in behavioral and physiological panic-associated responses, presence of chemosensitive targets in this region is of interest. However, additional studies are required to determine the involvement of this proposed PAG suffocation alarm in PD.
Translational relevance of acid-chemosensory mechanisms: from animal models to PD
Findings in translational rodent models of panic have provided information on potential target receptors and ion channels, as well as brain areas that may contribute to the pathophysiology of panic in humans (Figure 2). Clinical studies over the years have shown that a problem with acid–base homeostasis may exist in PD subjects pointing to the relevance of pH sensing and transduction targets as well as underlying circuits that contribute to pathophysiologic responses. For some acid chemosensors such as the ASIC1 channels, evidence from preclinical work translates well to human PD. Genetic studies have shown an association of polymorphisms in the ASIC-1a gene with PD.105 However, regional attributes and circuits underlying ASIC contributions to PD are less clear. A role of ASIC1 in the amygdala function is further supported in a rodent CO2 studies; however, it is noteworthy that Urbach–Wiethe subjects with damaged amygdala elicited panic and fear response to CO2 supporting the relevance of exrtra-amygdalar chemosensory in interoceptive threat detection and translation into panic. Here again, translational studies have suggested that dorsomedial hypothalamus, PAG, brain stem raphe and CVOs are potential sites of interest to PD. In some cases, findings from separate studies converge on selective sites that may be of particular relevance to PD. For example, studies on sodium lactate infusion in rodents by Shekhar’s group highlight an important role of CVOs, such as the subfornical organ and organum vasculosum of the lamina terminalis as sites for initiation of panic responses evoked by lactate.118 We recently demonstrated abundance of an acid-sensing TDAG8 receptor in these areas that are recruited in panic responses to CO2 inhalation, a panicogen.108, 141 Thus, areas devoid of a blood–brain barrier that can sense central and systemic homeostatic mileau may represent upstream detection sites for panic initiation especially given their connectivity to downstream sites for expression of behavioral and physiological responses as well as the forebrain regions such as the insula that have been implicated in PD. Acid chemosensors on orexin and 5-HT neurons provides further insights into integration of interoceptive pH fluctuations leading to behavioral and respiratory arousal. Association between polymorphisms in the TPH-2 gene and susceptibility to panic symptoms evoked by CO2 inhalation in humans,96 suggests the potential convergence of serotoninergic and CO2 chemosensory mechanisms. Future studies focusing on the crosstalk between exteroceptive and interoceptive pathways and mechanisms will be crucial to fully appreciate the unique pathophysiology of PD.
Potential therapeutic agents targeting pH chemosensory mechanisms
A role for pH and chemosensory mechanisms in panic physiology is increasingly appreciated on the basis of the recent explosion of data from preclinical studies; however, the translation to humans remains somewhat unclear and the degree to which these systems may be targeted by psychopharmacologic interventions is relatively unexplored. Thus, the lack of investigations related to pH or chemosensory modulating therapeutics is of high public health significance given that therapeutic options for PD are limited. As described above, selective serotonin reuptake inhibitors are the most commonly prescribed medications and, although they are effective in some patients, there are numerous side effects, delays in onset of action, whereas other modalities such as benzodiazepines—which have rapid onset of action—are associated with distinct limitations, including the risk of dependence and withdrawal. To date, there have been no clinical studies in PD patients on interventions targeted towards control of pH imbalance or chemosensory ion channel or receptor blockers. It is evident that antagonists for chemosensors such as ASIC1 ion channel may be a promising therapeutic target for PD. ASIC ion channel antagonist, such as amiloride have held promise for other acidosis associated conditions such as pain, stroke, migraine, spinal cord injury and multiple sclerosis (reviewed in Wemmie148). Recent studies show neuroprotective effects of amiloride in patients with progressive multiple sclerosis,149 supporting the safety and efficacy of this agent for alleviating pH-associated pathophysiology. Another promising target for PD are orexin receptor antagonists. Pre-treating with Orexin-1 receptor antagonist, SB334867, attenuates anxiety-like responses to CO2 inhalation in rats.124 In addition, silencing ORX precursor gene expression in the dorsal medial-perifornical hypothalamus/perifornical region, or systemically pretreating rats with SB334867 blocked intravenous sodium lactate-induced anxiety-related behavior and cardioexcitation.119 SB334867 also attenuated anxiety and panic-relevant behaviors induced by benzodiazepine inverse agonist FG-7142 and adenosine receptor antagonist, caffeine suggesting that orexin antagonists act on pathways common to diverse triggers of panic.115 In view of the relevance of acid–base dysregulation and CO2 sensitivity to panic it would be of interest to discuss studies on carbonic anhydrase inhibitor, acetazolamide in panic patients. Acetazolamide blocks the facilitated conversion of carbon dioxide to carbonic acid and finally bicarbonate and hydrogen ions, leading to a significant increase in CO2 concentration. It was hypothesized that acetazolamide would induce panic attacks in PD patients due to a rise in CO2 concentration. Contrary to this, panic attacks were not induced by acetazolamide.53, 150 In fact, it was suggested that acetazolamide may serve as an anti-panic agent, as it buffers against CO2-induced hydrogen ion fluctuations.151 As noted in that study, acetazolamide prevents the accumulation of H+ ions due to inhibition of carbonic anhydrase and provides support for H+ ions as the primary biological effector in CO2-evoked panic. These studies warrant further investigation and support for the therapeutic potential of acetazolamide in PD.
Conclusions and future directions
It is clear from the clinical and preclinical findings discussed in preceding sections that interoceptive acid/base imbalance and pH chemosensory mechanisms may contribute to certain aspects of PD, particularly uncued panic attacks. Converging evidence from neuroimaging, genetics and rodent preclinical models strongly supports that underlying abnormalities in pH homeostasis and chemosensation may be an important causative factor in panicogenesis.
However, it should be noted that, while there is significant evidence and consensus on the role of pH homeostasis and impaired acid–base buffering in patients with PD, not all data support the link between pH, specifically acidosis, and panic attacks. As described herein, clinical, genetic and preclinical studies on CO2 inhalation strongly support this link. Although not all panicogens (for example, sodium lactate and doxapram, which may cause respiratory distress, pH shifts or compensatory acidosis as a response to alkalosis) support the notion that acidosis provokes panic. In this regard, future studies are required to further clarify these inconsistencies.
Although pH sensing may contribute to panic, systems regulating stress, arousal, fear and anxiety are also relevant, particularly in the maintenance of the disorder. It is possible that panic vulnerability occurs due to a combined deficit in the processing of internal and external threats. The late onset of PD also suggests that more than one phenomenon is required for the manifestation of the disorder. An interesting future research direction would be to study the connectivity and crosstalk between pH chemosensory mechanisms and exteroceptive threat response systems (see Figure 2). There is also a need for the development of preclinical animal models where stress and pH chemosensory threat processing and translation should be simulated as this scenario is more likely to occur in humans. These models will also be relevant for therapeutic testing of novel agents. Another important area would be to study the interaction and communication between different pH chemosensory molecules in the brain. The presence of multiple sensory mechanisms at distinct sites is reflective of a highly sensitive pH threat detection system functioning at different thresholds and sensitivities, which may be relevant to PD. In conclusion, pH homeostasis and chemosensation remains an important area of investigation that furthers our understanding of panic pathophysiology and treatment.
References
Grant BF, Hasin DS, Stinson FS, Dawson DA, Goldstein RB, Smith S et al. The epidemiology of DSM-IV panic disorder and agoraphobia in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry 2006; 67: 363–374.
Beesdo K, Pine DS, Lieb R, Wittchen H-U . Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry 2010; 67: 47–57.
Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE . Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 2005; 62: 617–627.
Gadermann AM, Alonso J, Vilagut G, Zaslavsky AM, Kessler RC . Comorbidity and disease burden in the National Comorbidity Survey Replication (NCS-R). Depress Anxiety 2012; 29: 797–806.
Brown LA, Gaudiano BA, Miller IW . The impact of panic-agoraphobic comorbidity on suicidality in hospitalized patients with major depression. Depress Anxiety 2010; 27: 310–315.
Wang PS, Lane M, Olfson M, Pincus HA, Wells KB, Kessler RC . Twelve-month use of mental health services in the United States: results from the National Comorbidity Survey Replication. Arch Gen Psychiatry 2005; 62: 629–640.
Furukawa TA, Watanabe N, Churchill R . Combined psychotherapy plus antidepressants for panic disorder with or without agoraphobia. Cochrane Database Syst Rev 2007; CD004364.
Watanabe N, Churchill R, Furukawa TA . Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev 2009; CD005335.
Association AP Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. American Psychiatric Association: Arlington, VA, USA, 2013.
Graeff FG, Del-Ben CM . Neurobiology of panic disorder: from animal models to brain neuroimaging. Neurosci Biobehav Rev 2008; 32: 1326–1335.
Papp L a, Klein DF, Gorman JM . Carbon dioxide hypersensitivity, hyperventilation, and panic disorder. Am J Psychiatry 1993; 150: 1149–1157.
Gorman JM, Kent JM, Sullivan GM, Coplan JD . Neuroanatomical hypothesis of panic disorder, revised. Am J Psychiatry 2000; 157: 493–505.
Dresler T, Guhn A, Tupak S V, Ehlis A-C, Herrmann MJ, Fallgatter AJ et al. Revise the revised? New dimensions of the neuroanatomical hypothesis of panic disorder. J Neural Transm 2013; 120: 3–29.
Johnson PL, Federici LM, Shekhar A . Etiology, triggers and neurochemical circuits associated with unexpected, expected, and laboratory-induced panic attacks. Neurosci Biobehav Rev 2014; 46P3: 429–454.
Wemmie JA . Neurobiology of panic and pH chemosensation in the brain. Dialogues Clin Neurosci 2011; 13: 475–483.
Esquivel G, Schruers KR, Maddock RJ, Colasanti A, Griez EJ, KR S et al. Acids in the brain: a factor in panic? J Psychopharmacol 2010; 24: 639–647.
Maddock RJ . The lactic acid response to alkalosis in panic disorder: an integrative re view. J Neuropsychiatry Clin Neurosci 2001; 13: 22–34.
James W . The Principles of Psychology. Holt H: New York, NY, USA, 1890.
Meuret AE, Rosenfield D, Wilhelm FH, Zhou E, Conrad A, Ritz T et al. Do unexpected panic attacks occur spontaneously? Biol Psychiatry 2011; 70: 985–991.
Nattie E . Central chemoreceptors, pH and respiratory control. In: Kaila K, Ransom BR (eds). pH and Brain Function. Wiley-Liss: Wilmington, DE, USA, 1998; pp 535–560.
Sikter A, Frecska E, Braun I . The role of hyperventilation: hypocapnia in the pathomechanism of panic disorder. Rev Bras Psiquiatr 2007; 29: 375–379.
Dager SR, Strauss WL, Marro KI, Richards TL, Metzger GD, Artru AA . Proton magnetic resonance spectroscopy investigation of hyperventilation in subjects with panic disorder and comparison subjects. Am J Psychiatry 1995; 152: 666–672.
Maddock RJ, Buonocore MH, Copeland LE, Richards AL . Elevated brain lactate responses to neural activation in panic disorder: a dynamic 1H-MRS study. Mol Psychiatry 2008; 14: 537–545.
Ueda Y, Aizawa M, Takahashi A, Fujii M, Isaka Y . Exaggerated compensatory response to acute respiratory alkalosis in panic disorder is induced by increased lactic acid production. Nephrol Dial Transplant 2009; 24: 825–828.
Colasanti A, Salamon E, Schruers K, van Diest R, van Duinen M, Griez EJ . Carbon dioxide-induced emotion and respiratory symptoms in healthy volunteers. Neuropsychopharmacology 2008; 33: 3103–3110.
Ramirez J-M . The integrative role of the sigh in psychology, physiology, pathology, and neurobiology. Prog Brain Res 2014; 209: 91–129.
Klein DF False . suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch Gen Psychiatry 1993; 50: 306–317.
Maddock RJ, Buonocore MH, Miller AR, Yoon JH, Soosman SK, Unruh AM . Abnormal activity-dependent brain lactate and glutamate+glutamine responses in panic disorder. Biol Psychiatry 2013; 73: 1111–1119.
Dager SR, Friedman SD, Heide A, Layton ME, Richards T, Artru A et al. Two-dimensional proton echo-planar spectroscopic imaging of brain metabolic changes during lactate-induced panic. Arch Gen Psychiatry 1999; 56: 70–77.
Dager SR, Richards T, Strauss W, Artru A . Single-voxel 1H-MRS investigation of brain metabolic changes during lactate-induced panic. Psychiatry Res 1997; 76: 89–99.
Damasio A, Carvalho GB . The nature of feelings: evolutionary and neurobiological origins. Nat Rev Neurosci 2013; 14: 143–152.
Friedman SD, Mathis CM, Hayes C, Renshaw P, Dager SR . Brain pH response to hyperventilation in panic disorder: preliminary evidence for altered acid-base regulation. Am J Psychiatry 2006; 163: 710–715.
Goossens L, Leibold N, Peeters R, Esquivel G, Knuts I, Backes W et al. Brainstem response to hypercapnia: a symptom provocation study into the pathophysiology of panic disorder. J Psychopharmacol 2014; 28: 449–456.
Magnotta VA, Heo H-YY, Dlouhy BJ, Dahdaleh NS, Follmer RL, Thedens DR et al. Detecting activity-evoked pH changes in human brain. Proc Natl Acad Sci USA 2012; 109: 8270–8273.
Magnotta VA, Johnson CP, Follmer R, Wemmie JA . Functional t1ρ imaging in panic disorder. Biol Psychiatry 2014; 75: 884–891.
Rassovsky Y, Kushner MG . Carbon dioxide in the study of panic disorder: Issues of definition, methodology, and outcome. J Anxiety Disord 2003; 17: 1–32.
Cohen ME, White PD . Life situations, emotions, and neurocirculatory asthenia (anxiety neurosis, neurasthenia, effort syndrome). Psychosom Med 1951; 13: 335–357.
Pitts FN, McClure JN, Pitts FN Jr, McClure JN Jr . Lactate metabolism in anxiety neurosis. N Engl J Med 1967; 277: 1329–1336.
Lee Y-LJ, Curtis GC, Weg JG, Abelson JL, Modell JG, Campbell KM . Panic attacks induced by doxapram. Biol Psychiatry 1993; 33: 295–297.
Balon R, Yeragani VK, Pohl R, Muench J, Berchou R . Somatic and psychological symptoms during isoproterenol-induced panic attacks. Psychiatry Res 1990; 32: 103–112.
Vilarim MM, Rocha Araujo DM, Nardi AE . Caffeine challenge test and panic disorder: a systematic literature review. Expert Rev Neurother 2011; 11: 1185–1195.
Griez E, Schruers K . Experimental pathophysiology of panic. J Psychosom Res 1998; 45: 493–503.
De Montigny C . Cholecystokinin tetrapeptide induces panic-like attacks in healthy volunteers. Preliminary findings. Arch Gen Psychiatry 1989; 46: 511–517.
Nutt DJ, Glue P, Lawson C, Wilson S . Flumazenil provocation of panic attacks. Evidence for altered benzodiazepine receptor sensitivity in panic disorder. Arch Gen Psychiatry 1990; 47: 917–925.
Charney DS, Woods SW, Goodman WK, Heninger GR . Serotonin function in anxiety: II. Effects of the serotonin agonist MCPP in panic disorder patients and healthy subjects. Psychopharmacology (Berl) 1987; 92: 14–24.
Maremmani I, Marini G, Fornai F . Naltrexone-induced panic attacks. Am J Psychiatry 1998; 155: 447.
Kent JM, Papp LA, Martinez JM, Browne ST, Coplan JD, Klein DF et al. Specificity of panic response to CO(2) inhalation in panic disorder: a comparison with major depression and premenstrual dysphoric disorder. Am J Psychiatry 2001; 158: 58–67.
Talesnik B, Berzak E, Ben-Zion I, Kaplan Z, Benjamin J . Sensitivity to carbon dioxide in drug-naïve subjects with post-traumatic stress disorder. J Psychiatr Res 2007; 41: 451–454.
Do Amaral JMX, Spadaro PTM, Pereira VM, de Oliveira e Silva AC, Nardi AE . The carbon dioxide challenge test in panic disorder: a systematic review of preclinical and clinical research. Rev Bras Psiquiatr 2013; 35: 318–331.
Leibold NK, Viechtbauer W, Goossens L, De Cort K, Griez EJ, Myin-Germeys I et al. Carbon dioxide inhalation as a human experimental model of panic: the relationship between emotions and cardiovascular physiology. Biol Psychol 2013; 94: 331–340.
Huckstepp RTR, Dale N . Redefining the components of central CO2 chemosensitivity—towards a better understanding of mechanism. J Physiol 2011; 589: 5561–5579.
Sanderson WC, Wetzler S . Five percent carbon dioxide challenge: Valid analogue and marker of panic disorder? Biol Psychiatry 1990; 27: 689–701.
Mathew RJ, Wilson WH, Tant S . Responses to hypercarbia induced by acetazolamide in panic disorder patients. Am J Psychiatry 1989; 146: 996–1000.
Gorman JM, Papp LA, Coplan J, Martinez J, Liebowitz MR, Klein DF . The effect of acetazolamide on ventilation in panic disorder patients. Am J Psychiatry 1993; 150: 1480–1484.
Kelly MM, Forsyth JP, Karekla M . Sex differences in response to a panicogenic challenge procedure: an experimental evaluation of panic vulnerability in a non-clinical sample. Behav Res Ther 2006; 44: 1421–1430.
Bunaciu L, Feldner MT, Babson K a, Zvolensky MJ, Eifert GH . Biological sex and panic-relevant anxious reactivity to abrupt increases in bodily arousal as a function of biological challenge intensity. J Behav Ther Exp Psychiatry 2012; 43: 526–531.
Nillni YI, Berenz EC, Rohan KJ, Zvolensky MJ . Sex differences in panic-relevant responding to a 10% carbon dioxide-enriched air biological challenge. J Anxiety Disord 2012; 26: 165–172.
Nardi AE, Valença AM, Lopes FL, de-Melo-Neto VL, Freire RC, Veras AB et al. Caffeine and 35% carbon dioxide challenge tests in panic disorder. Hum Psychopharmacol 2007; 22: 231–240.
Griez EJ, Colasanti A, van Diest R, Salamon E, Schruers K . Carbon dioxide inhalation induces dose-dependent and age-related negative affectivity. PLoS One 2007; 2: e987.
Van den Hout MA, van der Molen GM, Griez E, Lousberg H, Nansen A . Reduction of CO2-induced anxiety in patients with panic attacks after repeated CO2 exposure. Am J Psychiatry 1987; 144: 788–791.
Prenoveau JM, Forsyth JP, Kelly MM, Barrios V . Repeated exposure to 20% CO2 challenge and risk for developing panic attacks: a controlled 6- and 12-month follow-up in a nonclinical sample. J Anxiety Disord 2006; 20: 1158–1167.
Perna G, Bertani A, Caldirola D, Gabriele A, Cocchi S, Bellodi L . Antipanic drug modulation of 35% CO2 hyperreactivity and short-term treatment outcome. J Clin Psychopharmacol 2002; 22: 300–308.
Bailey JE, Papadopoulos A, Seddon K, Nutt DJ . A comparison of the effects of a subtype selective and non-selective benzodiazepine receptor agonist in two CO(2) models of experimental human anxiety. J Psychopharmacol 2009; 23: 117–122.
Bailey JE, Papadopoulos A, Diaper A, Phillips S, Schmidt M, van der Ark P et al. Preliminary evidence of anxiolytic effects of the CRF(1) receptor antagonist R317573 in the 7.5% CO(2) proof-of-concept experimental model of human anxiety. J Psychopharmacol 2011; 25: 1199–1206.
Poma SZ, Merlo-Pich E, Bettica P, Bani M, Fina P, Ziviani L et al. Anxiolytic effects of vestipitant in a sub-group of healthy volunteers known to be sensitive to CO2 challenge. J Psychopharmacol 2014; 28: 491–497.
Gorman JM, Battista D, Goetz RR, Dillon DJ, Liebowitz MR, Fyer AJ et al. A comparison of sodium bicarbonate and sodium lactate infusion in the induction of panic attacks. Arch Gen Psychiatry 1989; 46: 145–150.
Goetz RR, Klein DF, Gorman JM . Consistencies between recalled panic and lactate-induced panic. Anxiety 1994; 1: 31–36.
Carr DB, Sheehan D V, Surman OS, Coleman JH, Greenblatt DJ, Heninger GR et al. Neuroendocrine correlates of lactate-induced anxiety and their response to chronic alprazolam therapy. Am J Psychiatry 1986; 143: 483–494.
Cowley DS, Dager SR, Roy-Byrne PP, Avery DH, Dunner DL . Lactate vulnerability after alprazolam versus placebo treatment of panic disorder. Biol Psychiatry 1991; 30: 49–56.
Rifkin A, Klein DF, Dillon D, Levitt M . Blockade by imipramine or desipramine of panic induced by sodium lactate. Am J Psychiatry 1981; 138: 676–677.
Tsacopoulos M, Magistretti PJ . Metabolic coupling between glia and neurons. J Neurosci 1996; 16: 877–885.
Van Hall G, Strømstad M, Rasmussen P, Jans O, Zaar M, Gam C et al. Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab 2009; 29: 1121–1129.
Dager SR, Rainey JM, Kenny MA, Artru AA, Metzger GD, Bowden DM . Central nervous system effects of lactate infusion in primates. Biol Psychiatry 1990; 27: 193–204.
Peskind ER, Jensen CF, Pascualy M, Tsuang D, Cowley D, Martin DC et al. Sodium lactate and hypertonic sodium chloride induce equivalent panic incidence, panic symptoms, and hypernatremia in panic disorder. Biol Psychiatry 1998; 44: 1007–1016.
Seier FE, Kellner M, Yassouridis A, Heese R, Strian F, Wiedemann K . Autonomic reactivity and hormonal secretion in lactate-induced panic attacks. Am J Physiol 1997; 272: H2630–H2638.
Layton ME, Friedman SD, Dager SR . Brain metabolic changes during lactate-induced panic: Effects of gabapentin treatment. Depress Anxiety 2001; 14: 251–254.
Ströhle A, Romeo E, di Michele F, Pasini A, Hermann B, Gajewsky G et al. Induced panic attacks shift gamma-aminobutyric acid type A receptor modulatory neuroactive steroid composition in patients with panic disorder: preliminary results. Arch Gen Psychiatry 2003; 60: 161–168.
Ward JW, Franko BV . A new centrally acting agent (AHR-619) with marked respiratory stimulant, pressor and awakening effects. Fed Proc 1962; 21: 325.
Burki NK . Ventilatory effects of doxapram in conscious human subjects. Chest 1984; 85: 600–604.
Gutman DA, Coplan J, Papp L, Martinez J, Gorman J . Doxapram-induced panic attacks and cortisol elevation. Psychiatry Res 2005; 133: 253–261.
Abelson JL, Nesse RM, Weg JG, Curtis GC . Respiratory psychophysiology and anxiety: cognitive intervention in the doxapram model of panic. Psychosom Med 1996; 58: 302–313.
Garakani A, Buchsbaum MS, Newmark RE, Goodman C, Aaronson CJ, Martinez JM et al. The effect of doxapram on brain imaging in patients with panic disorder. Eur Neuropsychopharmacol 2007; 17: 672–686.
Cotten JF, Keshavaprasad B, Laster MJ, Eger EI II, Yost CS . The ventilatory stimulant doxapram inhibits TASK tandem pore (K 2 P) potassium channel function but does not affect minimum alveolar anesthetic concentration. Anesth Analg 2006; 102: 779–785.
Washburn CP, Sirois JE, Talley EM, Guyenet PG, Bayliss DA . Serotonergic raphe neurons express TASK channel transcripts and a TASK-like pH- and halothane-sensitive K+ conductance. J Neurosci 2002; 22: 1256–1265.
Yost CS . A new look at the respiratory stimulant doxapram. CNS Drug Rev 2006; 12: 236–249.
Pauls DL, Noyes R, Crowe RR . The familial prevalence in second-degree relatives of patients with anxiety neurosis (panic disorder). J Affect Disord 1979; 1: 279–285.
Perna G, Cocchi S, Bertani A, Arancio C, Bellodi L . Sensitivity to 35% CO2 in healthy first-degree relatives of patients with panic disorder. Am J Psychiatry 1995; 152: 623–625.
Coryell W . Hypersensitivity to carbon dioxide as a disease-specific trait marker. Biol Psychiatry 1997; 41: 259–263.
Bellodi L, Perna G, Caldirola D, Arancio C, Bertani A, Di Bella D . CO2-induced panic attacks: a twin study. Am J Psychiatry 1998; 155: 1184–1188.
Battaglia M, Pesenti-Gritti P, Spatola CAM, Ogliari A, Tambs K . A twin study of the common vulnerability between heightened sensitivity to hypercapnia and panic disorder. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 586–593.
Roberson-Nay R, Berenz EC, Acierno R, Tran TL, Trung LT, Tam NT et al. Characteristics of individuals meeting criteria for new onset panic attacks following exposure to a typhoon. Psychiatry Res 2013; 209: 574–578.
Battaglia M, Pesenti-Gritti P, Medland SE, Ogliari A, Tambs K, Spatola CAM . A genetically informed study of the association between childhood separation anxiety, sensitivity to CO(2), panic disorder, and the effect of childhood parental loss. Arch Gen Psychiatry 2009; 66: 64–71.
D’Amato FR, Zanettini C, Lampis V, Coccurello R, Pascucci T, Ventura R et al. Unstable maternal environment, separation anxiety, and heightened CO2 sensitivity induced by gene-by-environment interplay. PLoS One 2011; 6: e18637.
Roberson-Nay R, Klein DF, Klein RG, Mannuzza S, Moulton JL, Guardino M et al. Carbon dioxide hypersensitivity in separation-anxious offspring of parents with panic disorder. Biol Psychiatry 2010; 67: 1171–1177.
Battaglia M, Ogliari A, D’Amato F, Kinkead R . Early-life risk factors for panic and separation anxiety disorder: Insights and outstanding questions arising from human and animal studies of CO2 sensitivity. Neurosci Biobehav Rev 2014; 46P3: 455–464.
Abe R, Watanabe Y, Tachibana A, Nunokawa A, Shindo M, Hasegawa N et al. Exploration of a possible association between the tryptophan hydroxylase 2 (TPH2) gene and panic symptoms induced by carbon dioxide in healthy individuals. Psychiatry Res 2012; 197: 358–359.
Gyawali S, Subaran R, Weissman MM, Hershkowitz D, McKenna MC, Talati A et al. Association of a polyadenylation polymorphism in the serotonin transporter and panic disorder. Biol Psychiatry 2010; 67: 331–338.
Strug LJ, Suresh R, Fyer AJ, Talati A, Adams PB, Li W et al. Panic disorder is associated with the serotonin transporter gene (SLC6A4) but not the promoter region (5-HTTLPR). Mol Psychiatry 2010; 15: 166–176.
Domschke K, Braun M, Ohrmann P, Suslow T, Kugel H, Bauer J et al. Association of the functional -1019C/G 5-HT1A polymorphism with prefrontal cortex and amygdala activation measured with 3T fMRI in panic disorder. Int J Neuropsychopharmacol 2006; 9: 349–355.
Kim Y-K, Lee H-J, Yang J-C, Hwang J-A, Yoon H-K . A tryptophan hydroxylase 2 gene polymorphism is associated with panic disorder. Behav Genet 2009; 39: 170–175.
Perkins AM, Ettinger U, Williams SCR, Reuter M, Hennig J, Corr PJ . Flight behaviour in humans is intensified by a candidate genetic risk factor for panic disorder: evidence from a translational model of fear and anxiety. Mol Psychiatry 2011; 16: 242–244.
Paul ED, Johnson PL, Shekhar A, Lowry CA . The Deakin/Graeff hypothesis: focus on serotonergic inhibition of panic. Neurosci Biobehav Rev 2014; 46: 379–396.
Reschke AH, Mannuzza S, Chapman TF, Lipsitz JD, Liebowitz MR, Gorman JM et al. Sodium lactate response and familial risk for panic disorder. Am J Psychiatry 1995; 152: 277–279.
Philibert RA, Nelson JJ, Sandhu HK, Crowe RR, Coryell WH . Association of an exonic LDHA polymorphism with altered respiratory response in probands at high risk for panic disorder. Am J Med Genet B Neuropsychiatr Genet 2003; 117B: 11–17.
Smoller JW, Gallagher PJ, Duncan LE, McGrath LM, Haddad SA, Holmes AJ et al. The human ortholog of acid-sensing ion channel gene ASIC1a is associated with panic disorder and amygdala structure and function. Biol Psychiatry 2014; 76: 902–910.
Putnam RW . CO2 chemoreception in cardiorespiratory control. J Appl Physiol 2010; 108: 1796–1802.
Kinkead R, Tenorio L, Drolet G, Bretzner F, Gargaglioni L . Respiratory manifestations of panic disorder in animals and humans: a unique opportunity to understand how supramedullary structures regulate breathing. Respir Physiol Neurobiol 2014; 204: 3–13.
Vollmer LE, Rush J, Li K-Y, Lewkowich I, Putnam R, Sah R . Microglial acid-sensing T-Cell Death Associated Gene-8 (TDAG8) receptor in CO2 evoked behavior and physiology: relevance to panic. In: Society for Neuroscience Abstracts 2012.
Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH, Welsh MJ . Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci 2003; 23: 5496–5502.
Cardinal RN, Parkinson JA, Hall J, Everitt BJ . Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev 2002; 26: 321–352.
Ziemann AE, Allen JE, Dahdaleh NS, Drebot II, Coryell MW, Wunsch AM et al. The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell 2009; 139: 1012–1021.
Coryell MW, Ziemann AE, Westmoreland PJ, Haenfler JM, Kurjakovic Z, Zha X et al. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry 2007; 62: 1140–1148.
Feinstein JS, Buzza C, Hurlemann R, Follmer RL, Dahdaleh NS, Coryell WH et al. Fear and panic in humans with bilateral amygdala damage. Nat Neurosci 2013; 16: 270–272.
Feinstein JS, Adolphs R, Damasio A, Tranel D . The human amygdala and the induction and experience of fear. Curr Biol 2011; 21: 34–38.
Johnson PL, Shekhar A . An animal model of panic vulnerability with chronic disinhibition of the dorsomedial/perifornical hypothalamus. Physiol Behav 2012; 107: 686–698.
Shekhar A, Keim SRR, Simon JRR, McBride WJJ . Dorsomedial hypothalamic GABA dysfunction produces physiological arousal following sodium lactate infusions. Pharmacol Biochem Behav 1996; 55: 249–256.
Wilent WB, Oh MY, Buetefisch CM, Bailes JE, Cantella D, Angle C et al. Induction of panic attack by stimulation of the ventromedial hypothalamus. J Neurosurg 2010; 112: 1295–1298.
Shekhar A, Keim SR . The circumventricular organs form a potential neural pathway for lactate sensitivity: implications for panic disorder. J Neurosci 1997; 17: 9726–9735.
Johnson PL, Truitt W, Fitz SD, Minick PE, Dietrich A, Sanghani S et al. A key role for orexin in panic anxiety. Nat Med 2010; 16: 111–115.
Shekhar A, Johnson PL, Sajdyk TJ, Fitz SD, Keim SR, Kelley PE et al. Angiotensin-II is a putative neurotransmitter in lactate-induced panic-like responses in rats with disruption of GABAergic inhibition in the dorsomedial hypothalamus. J Neurosci 2006; 26: 9205–9215.
Fukuda Y, Sato A, Suzuki A, Trzebski A . Autonomic nerve and cardiovascular responses to changing blood oxygen and carbon dioxide levels in the rat. J Auton Nerv Syst 1989; 28: 61–74.
De Lecea L, Kilduff TS, Peyron C, Gao X-B, Foye PE, Danielson PE et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 1998; 95: 322–327.
Deng B-S, Nakamura A, Zhang W, Yanagisawa M, Fukuda Y, Kuwaki T . Contribution of orexin in hypercapnic chemoreflex: evidence from genetic and pharmacological disruption and supplementation studies in mice. J Appl Physiol 2007; 103: 1772–1779.
Johnson PL, Samuels BC, Fitz SD, Lightman SL, Lowry CA, Shekhar A . Activation of the orexin 1 receptor is a critical component of CO2-mediated anxiety and hypertension but not bradycardia. Neuropsychopharmacology 2012; 37: 1911–1922.
Yeoh JW, Campbell EJ, James MH, Graham BA, Dayas C V . Orexin antagonists for neuropsychiatric disease: progress and potential pitfalls. Front Neurosci 2014; 8: 36.
Richerson GB . Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci 2004; 5: 449–461.
Severson C a, Wang W, Pieribone V a, Dohle CI, Richerson GB . Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci 2003; 6: 1139–1140.
Bradley SR, Pieribone VA, Wang W, Severson CA, Jacobs RA, Richerson GB . Chemosensitive serotonergic neurons are closely associated with large medullary arteries. Nat Neurosci 2002; 5: 401–402.
Buchanan GF, Richerson GB . Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol 2009; 167: 9–19.
Iceman KE, Harris MB . A group of non-serotonergic cells is CO2-stimulated in the medullary raphé. Neuroscience 2014; 259: 203–213.
Colasanti A, Esquivel G, den Boer E, Horlings A, Dandachi A, Oostwegel JL et al. Effects of tryptophan depletion and tryptophan loading on the affective response to high-dose CO2 challenge in healthy volunteers. Psychopharmacology (Berl) 2011; 215: 739–748.
Choi JW, Lee SY, Choi Y . Identification of a putative G protein-coupled receptor induced during activation-induced apoptosis of T cells. Cell Immunol 1996; 168: 78–84.
Kyaw H, Zeng Z, Su K, Fan P, Shell BK, Carter KC et al. Cloning, characterization, and mapping of human homolog of mouse T-cell death-associated gene. DNA Cell Biol 1998; 17: 493–500.
McGuire J, Herman JP, Ghosal S, Eaton K, Sallee FR, Sah R et al. Acid-sensing by the T cell death-associated gene 8 (TDAG8) receptor cloned from rat brain. Biochem Biophys Res Commun 2009; 386: 420–425.
Ishii S, Kihara Y, Shimizu T . Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J Biol Chem 2005; 280: 9083–9087.
Ludwig M, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U et al. Proton-sensing G-protein-coupled receptors. Nature 2003; 425: 93–98.
Radu CG, Nijagal A, McLaughlin J, Wang L, Witte ON . Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci USA 2005; 102: 1632–1637.
Wang J-Q, Kon J, Mogi C, Tobo M, Damirin A, Sato K et al. TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J Biol Chem 2004; 279: 45626–45633.
Johnson AK, Gross PM . Sensory circumventricular organs and brain homeostatic pathways. FASEB J 1993; 7: 678–686.
Sisó S, Jeffrey M, González L . Sensory circumventricular organs in health and disease. Acta Neuropathol 2010; 120: 1–17.
Vollmer LE, Ghosal S, Li K-Y, Putnam R, Sah R . Acid Sensing T-Cell Death Associated Gene-8 (TDAG8) Receptor in CO2 Evoked Fear: Relevance to Panic Pathophysiology. In: Society for Neroscience Abstracts 2011.
Schenberg LC, Bittencourt a S, Sudré EC, Vargas LC . Modeling panic attacks. Neurosci Biobehav Rev 2001; 25: 647–659.
Moreira FA, Gobira PH, Viana TG, Vicente MA, Zangrossi H, Graeff FG . Modeling panic disorder in rodents. Cell Tissue Res 2013; 354: 119–125.
Nashold BS, Wilson WP, Slaughter DG . Sensations evoked by stimulation in the midbrain of man. J Neurosurg 1969; 30: 14–24.
Protopopescu X, Pan H, Tuescher O, Cloitre M, Goldstein M, Engelien A et al. Increased brainstem volume in panic disorder: a voxel-based morphometric study. Neuroreport 2006; 17: 361–363.
Uchida RR, Del-Ben CM, Busatto GF, Duran FLS, Guimarães FS, Crippa JAS et al. Regional gray matter abnormalities in panic disorder: a voxel-based morphometry study. Psychiatry Res 2008; 163: 21–29.
Schimitel FG, de Almeida GM, Pitol DN, Armini RS, Tufik S, Schenberg LC . Evidence of a suffocation alarm system within the periaqueductal gray matter of the rat. Neuroscience 2012; 200: 59–73.
Wemmie JA, Taugher RJ, Kreple CJ . Acid-sensing ion channels in pain and disease. Nat Rev Neurosci 2013; 14: 461–471.
Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MI et al. Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain 2013; 136: 106–115.
Klein DF, Papp LA, Gorman JM . Acetazolamide as a control for CO2-induced panic. Am J Psychiatry 1990; 147: 677–678.
Klein DF . Testing the suffocation false alarm theory of panic disorder. Anxiety 1994; 1: 1–7.
Acknowledgements
We acknowledge support from the National Institute of Mental Health (MS) to JRS and RS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JRS receives or has received research support from Edgemont, Eli Lilly, Shire, Forest Research Laboratories, Lundbeck, the American Academy of Child and Adolescent Psychiatry and the National Institute of Mental Health, and is an employee of the University of Cincinnati. He receives royalties from Springer Publishing for two books. RS is a consultant for Ono Pharmaceuticals, Japan. The remaining author declares no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vollmer, L., Strawn, J. & Sah, R. Acid–base dysregulation and chemosensory mechanisms in panic disorder: a translational update. Transl Psychiatry 5, e572 (2015). https://doi.org/10.1038/tp.2015.67
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.67
