
Abstract
Response to treatment with selective serotonin reuptake inhibitors (SSRIs) varies considerably between patients. The International SSRI Pharmacogenomics Consortium (ISPC) was formed with the primary goal of identifying genetic variation that may contribute to response to SSRI treatment of major depressive disorder. A genome-wide association study of 4-week treatment outcomes, measured using the 17-item Hamilton Rating Scale for Depression (HRSD-17), was performed using data from 865 subjects from seven sites. The primary outcomes were percent change in HRSD-17 score and response, defined as at least 50% reduction in HRSD-17. Data from two prior studies, the Pharmacogenomics Research Network Antidepressant Medication Pharmacogenomics Study (PGRN-AMPS) and the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, were used for replication, and a meta-analysis of the three studies was performed (N=2394). Although many top association signals in the ISPC analysis map to interesting candidate genes, none were significant at the genome-wide level and the associations were not replicated using PGRN-AMPS and STAR*D data. Top association results in the meta-analysis of response included single-nucleotide polymorphisms (SNPs) in the HPRTP4 (hypoxanthine phosphoribosyltransferase pseudogene 4)/VSTM5 (V-set and transmembrane domain containing 5) region, which approached genome-wide significance (P=5.03E−08) and SNPs 5’ upstream of the neuregulin-1 gene, NRG1 (P=1.20E−06). NRG1 is involved in many aspects of brain development, including neuronal maturation and variations in this gene have been shown to be associated with increased risk for mental disorders, particularly schizophrenia. Replication and functional studies of these findings are warranted.
Similar content being viewed by others

Introduction
Major depressive disorder (MDD) is a serious psychiatric illness with a lifetime prevalence of ~13%.1 While several types of antidepressant medications have been shown to have beneficial effects for MDD symptoms, selection of the most appropriate medication for individual patients continues to be a challenge. Selective serotonin reuptake inhibitors (SSRIs) are the most commonly used medication class for MDD;2, 3 however, response to SSRI treatment varies considerably between patients, and it is widely recognized that identification of pharmacogenetic predictors of drug response has great potential to improve the treatment of MDD.4
Several genome-wide association studies (GWAS) of SSRI treatment outcomes have been performed.5, 6, 7, 8, 9 None of these studies have identified genetic variants that were associated with treatment outcomes at a genome-wide statistically significant level. Moreover, top findings from these studies have not been replicated in independent samples. Nevertheless, analysis of antidepressant response data using a mixed linear model approach to estimate the proportion of phenotypic variance explained by genome-wide single-nucleotide polymorphisms (SNPs, the GREML approach) implemented in the Genome-Wide Complex Trait Analysis package10 has demonstrated that common genetic variants explain a considerable proportion of individual differences in antidepressant response,11 providing compelling motivation for further pharmacogenomic studies.
Prior pharmacogenomic studies of antidepressant response have shown that GWAS have low power for discovery of relevant variants for this highly complex trait. Thus, large samples, such as those arising from the formation of collaborative consortia, will be necessary to make further progress in this field.12, 13 Taking this approach, Tansey et al.13 described a pharmacogenomic analysis of data arising from the NEWMEDS consortium, which includes response to serotonergic and noradrenergic antidepressants in over 2000 European-ancestry individuals with MDD. The analyses, which also included a meta-analysis with data from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study,14 did not identify any common genetic variants associated with antidepressant response at a genome-wide significant level. Another recent meta-analysis combined the results of three prior studies to search for genetic variation associated with remission following treatment with antidepressants, and with SSRIs (escitalopram or citalopram) specifically.12 These analyses failed to replicate the single-study results in the pooled analysis. However, the analysis of the entire sample (all antidepressants) resulted in one genome-wide significant association of a SNP located in an intronic region of the myosin X (MYO10) gene. The analysis of the SSRI-treated subset revealed an association between early SSRI response (within 2 weeks of treatment) and a SNP in an intergenic region on chromosome 5.
The International SSRI Pharmacogenomics Consortium (ISPC) was established to investigate the genetic factors contributing to variable response to SSRIs. Because a variety of SSRIs were used in the studies contributed to the consortium, this sample provides the opportunity to identify genes that contribute to the pharmacodynamic, though not pharmacokinetic, effects of these medications. This is the first report of findings from a GWAS of SSRI treatment response based on the ISPC sample. Data from two prior GWAS of SSRI response, the Mayo Clinic Pharmacogenomic Research Network Antidepressant Medication Pharmacogenomics Study (PGRN-AMPS)7, 15 and the STAR*D study,14 were used for replication analysis, and a meta-analysis of the three studies was performed.
Materials and methods
Study design and samples
The ISPC initially included eight member sites (Supplementary Table S1), with one site subsequently excluded because of unavailable clinical data. The seven studies included in the analyses represent five countries from Europe, North America and Asia, with 2/3 of the sample being Asian (Taiwan, Thailand and Japan) and 1/3 being of European descent (Germany and the United States). All sites included in analyses had approval to participate in the consortium from their local ethical review board.
In total, 1149 DNA samples and clinical data for 998 subjects were contributed. Demographic and clinical data provided by individual sites were curated (that is, collected, formatted and subjected to quality control) by staff at the Pharmacogenetics and Pharmacogenomics Knowledge Base (PharmGKB, www.pharmgkb.org). Supplementary Table S2 lists the collected data, which included information on factors previously shown to be associated with antidepressant therapy response. These data included demographic characteristics, socioeconomic data, depression history, co-occurring diseases, antidepressant medication, dose and compliance, use of concomitant medications, rating scores of the individual items of the Hamilton Rating Scale for Depression (HRSD) at baseline and follow-up visits, side effects (if reported) and possible dose or antidepressant change at follow-up visits. Several subjects that did not receive SSRIs during the study period were removed from analysis.
A description of each contributing study, including the medications used in the study, is included in Supplementary Table S1, and key features of the samples used in the analyses are summarized in Table 1. The pharmacogenomic analysis focused on treatment outcomes at 4 weeks, as this time point was common to all participating studies. Prior studies have shown that early response in depression is strongly correlated with late response, which supports our choice of 4 weeks of observation period.16, 17, 18 However, because 4 weeks is considered too early to observe complete remission, our analyses focused on percentage reduction in HRSD score and response (defined as ⩾50% reduction in HRSD score) rather than remission, as described in the Statistical analysis section.
Genotyping, quality control and imputation
All DNA samples (N=1149) were shipped to Mayo Clinic, Rochester, MN, USA for storage and plating, and were genotyped at the RIKEN Center of Integrative Medical Sciences (Yokohama, Japan) using Illumina HumanOmniExpressExome BeadChips. Three samples failed genotyping. Quality control assessments of the genetic data were performed as described in the Supplementary Materials, and structure analysis19 was used to infer ancestry using the genome-wide SNP data (Supplementary Figure S1), leading to removal of 16 subjects (11 related, 1 gender mismatch, 4 race outliers; see Supplementary Materials for details). As discussed in the Supplementary Methods section, there was clear population structure between sites, but without evidence of residual population stratification within sites. We verified that after adjustment for site, no further adjustment for ancestry via principal components was necessary. Thus, because site provided a good surrogate measure for ancestry, subsequent analyses were adjusted for site without further adjustment for population structure. After quality control, 631 765 genotyped SNPs remained for analysis. Genotypes at unobserved SNPs with minor allele frequency >0.01 in the reference population were imputed using Beagle (v3.3.1)20 with the 1000 genomes cosmopolitan reference panel. After removing markers with poor imputation quality (dosage R2<0.3), ~7 million imputed markers were available for analysis.
Statistical analysis
The pharmacogenomic analyses focused on treatment outcomes at 4 weeks, as this time point was common to all participating studies. GWA analyses were performed for two phenotypes: ‘% change in HRSD-17 score’ (%ΔHRSD defined as the change in HRSD-17 score divided by the baseline score) and ‘response’ (defined as ⩾50% reduction in HRSD-17 score from baseline to 4-week visit). Because response is defined by dichotomizing %ΔHRSD, these two outcomes are highly correlated, and therefore results of analyses of the two outcomes are also correlated. However, the Spearman correlation of the test-statistics (or equivalent P-values) from analyses of these two outcomes is not very high (0.50 in the ISPC data), indicating that analyses of these two correlated outcomes can detect associations with different SNPs. Thus, analyses of both outcomes are presented.
The analysis included subjects with HRSD-17 data at baseline and week 4 (one entire site was removed due to missing clinical information), and was restricted to subjects with a baseline HRSD-17 score ⩾10. Table 1 shows the demographic and clinical characteristics of the final sample used for the GWA analyses (865 subjects from seven sites). Distributions of baseline HRSD-17, sex and age differed significantly among sites. All GWA analyses were adjusted for age and sex, in addition to site. However, because age and sex were not statistically significant predictors of treatment outcomes after adjustment for site, we also repeated the GWA analyses without age and sex adjustment. The results were virtually identical, and only the age- and sex-adjusted results are presented.
For the GWA analysis of %ΔHRSD, the association of each SNP with the outcome was tested using a linear regression model, with the SNP (coded as 0, 1, 2) and the covariates (age, sex and site) as independent variables and %ΔHRSD as the dependent variable. Because the distribution of %ΔHRSD demonstrated departures from normality, a van der Warden transformation was applied before analysis. For the binary outcome (response), logistic regression was used to test for SNP effects, while accounting for the covariate effects.
Analyses were performed in R (http://www.R-project.org), SAS (SAS Institute, Cary, NC, USA) and PLINK.21 P-values <5 × 10−8 were considered statistically significant at the genome-wide level.
Replication of ISPC findings using PGRN-AMPS and STAR*D data
SNPs with the smallest P-values in each of the top 10 association regions in the two primary analyses (that is, top SNP in each region) were tested for association with equivalent outcomes in two independent samples: the PGRN-AMPS15 and STAR*D.14 GWA analyses utilizing these two samples are published,5, 7 and both data sets are available through controlled access via the database for Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap).
Both the PGRN-AMPS and STAR*D evaluated depression outcomes following treatment with SSRIs. Although the PGRN-AMPS utilized both the HRSD and the 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16) to measure depression symptoms at baseline and follow-up, STAR*D used primarily QIDS-C16 during the follow-up visits. Thus, replication analyses in the PGRN-AMPS data used the same treatment outcome definitions as the analysis of the ISPC data (based on HRSD-17), whereas replication analyses in the STAR*D data used equivalent %change and response outcomes definitions based on the QIDS-C16.
Analyses of the replication samples resembled those of the ISPC sample, using linear regression for quantitative outcomes and logistic regression for binary outcomes. Details of the quality control analyses in PGRN-AMPS have been described previously.7 To control for possible remaining population structure, analyses of the predominantly Caucasian PGRN-AMPS sample were adjusted for the first four eigenvectors derived from genome-wide SNP data thinned on the basis of linkage disequilibrium. After quality control filters (based on Garriock et al.,5 and removing 25 individuals from related pairs), genetic data were available for 1887 individuals from STAR*D; our analyses used data from 1529 subjects that had both baseline and 4-week QIDS-C16 measurements. Because the STAR*D sample included individuals of multiple ancestral backgrounds, analyses of this sample were adjusted for the first seven eigenvectors constructed from genome-wide SNP data, calculated in the same way as in the PGRN-AMPS.
Meta-analyses of ISPC, PGRN-AMPS and STAR*D data
We also performed GWA analyses on the combined ISPC+AMPS sample, followed by a fixed-effects meta-analysis to combine the ISPC+AMPS GWA results with STAR*D GWA results (total sample size of 2394). Estimated effect sizes from the two analyses were combined by averaging the estimates weighted by the inverse of their variances. The combined estimates were then tested by a two-sided Z-test using a pooled standard error.
Replication of results from prior genome-wide meta-analyses of antidepressant response
Finally, we used our sample to attempt replication of top findings from two recent large GWA studies of antidepressant response.12, 13 In particular, we attempted to replicate association with two SNPs that reached genome-wide significance in a published meta-analysis of GENDEP, MARS and STAR*D data12 (rs17651119 in MYO10, which was associated with 12-week percentage improvement in the entire sample, and the intergenic SNP rs12054895 that was associated with 2-week improvement in the SSRI-treated subset), as well as six other SNPs with suggestive evidence of association with one of the treatment outcomes (P<1.0E−06). Because this meta-analysis included the STAR*D sample, we evaluated replication of these prior findings in the ISPC sample and the combined ISPC plus PGRN-AMPS sample, but not in our meta-analysis that included STAR*D as that would not constitute an independent replication. We also investigated six SNPs with P<5.0E−06 reported by Tansey et al.13 for the overall analysis or the analysis of subjects treated with serotonergic antidepressants. For the SNPs reported by Tansey et al., we evaluated replication in our ISPC sample, as well as in our ISPC-AMPS-STAR*D meta-analysis.
Results
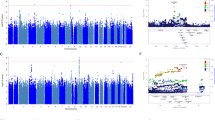
Out of 865 individuals with baseline and 4-week HRSD-17 scores (baseline mean (s.d.)=22.2 (5.5); 4-week mean (s.d.)=11.9 (6.1)), 416 (48%) were responders (Table 1). Figure 1 shows the Manhattan plots for the association of genotyped and imputed SNPs with the two clinical outcomes (%ΔHRSD and response), with the corresponding QQ plots shown in Supplementary Figure S2. The top 10 genotyped SNP associations (smallest P-values) for each outcome are listed in Table 2; none of these associations reached genome-wide significance. Top results for %ΔHRSD included SNPs in the genes MCPH1 (microcephalin 1) and STK39 (serine threonine kinase 39). Top results for response included MCPH1, PPIAP14 (peptidylprolyl isomerase A (cyclophilin A) pseudogene 14), SCN7A (sodium channel, type 7, alpha subunit), BRD2 (bromodomain containing 2) and HPRTP4 (hypoxanthine phosphoribosyltransferase pseudogene 4).

Manhattan plots showing genome-wide association results of the two outcome variables in the ISPC data analysis: (a) %ΔHRSD (b) response. HRSD, Hamilton Rating Scale for Depression; ISPC, International SSRI Pharmacogenomics Consortium.
The top 10 SNP association regions including imputed SNPs are shown in Supplementary Table S3, where for each association region, the SNP with the smallest P-value is listed. The top association signal in the analysis of imputed SNP data was a low frequency SNP, rs56058016, in the von Willebrand factor A domain containing 5B1 gene (VWA5B1) with P=1.1E−07 for association with %ΔHRSD. Other new signals in the analysis of imputed SNPs, not reaching genome-wide significance, included CTNNA3 (catenin alpha 3) for %ΔHRSD and RYR3 (ryanodine receptor 3) for response. None of the top associations were replicated in the PGRN-AMPS or STAR*D analyses (Supplementary Table S3). Replication of the top association signal in VWA5B1 could not be tested as this SNP was not imputed with adequate quality in the PGRN-AMPS and STAR*D data sets.
Results of the meta-analysis of the three data sets are shown in Figure 2 (Manhattan plots), Supplementary Figure S3 (QQ plots) and Table 3 (top 10 association SNPs for each outcome). In the meta-analysis of response, one SNP approached genome-wide significance (Table 3; rs2456568, P=5.0E−08). This SNP lies 3’ downstream of the pseudogene HPRTP4, with other SNPs in this region with P-values <10−6 being located 5’ upstream of the gene VSTM5 (V-set and transmembrane domain containing 5; Supplementary Figure S4). Other notable top association regions for response include the 5’ upstream region of the neuregulin gene, NRG1 (Figure 2b and Supplementary Figure S5). Top association signals (P<1.0E−06) for %change in depression score include SNPs in the MTMR12 (myotubularin-related protein 12) gene (Table 3).

Manhattan plots showing genome-wide association results for the two outcome variables in the meta-analysis of ISPC, AMPS and STAR*D data. (a) %Δ depression score, (b) response. AMPS, Antidepressant Medication Pharmacogenomics Study; ISPC, International SSRI Pharmacogenomics Consortium; STAR*D, Sequenced Treatment Alternatives to Relieve Depression.
Supplementary Table S4 shows results in the ISPC sample and the ISPC/PGRN-AMPS/STAR*D meta-analysis for SNPs selected on the basis of two recent large pharmacogenomic GWAS of antidepressant response.12, 13 Although the observed associations are not significant after correction for multiple testing of SNPs selected for these replication analyses, we observed marginally significant evidence of association of SNP rs11624702 in MDGA2 (MAM domain containing glycosylphosphatidylinositol anchor 2) with response in the ISPC alone (P=0.061) and in the combined ISPC-AMPS-STAR*D meta-analysis (P=0.029), with the minor allele being associated with lower odds of response (that is, worse clinical outcomes). Nominally significant association was also found for SNP rs7174755 in ITGA11 (integrin, alpha 11) in the combined ISPC+AMPS sample (P=0.010 for association with %ΔHRSD; P=0.025 for response). We also observed nominally significant association of the minor allele at SNP rs2546057 in KCTD15 (potassium channel tetramerization domain containing 15) with higher odds of response (better clinical outcome) in the ISPC sample, but this association did not remain significant in the combined ISPC+AMPS analysis.
Discussion
Genome-wide analyses of data from the ISPC presented here did not detect SNPs significantly associated with %ΔHRSD or response, and analyses of the PGRN-AMPS and STAR*D data did not replicate the top association signals from the analysis of the ISPC data. The top association signal was obtained for a set of imputed SNPs in the VWA5B1 gene (P=1.1E−07). This result should be interpreted cautiously as this is a rare imputed SNP. A meta-analysis that included the ISPC, PGRN-AMPS and STAR*D data revealed a SNP in the region of the HPRTP4 and VSTM5 genes that approached genome-wide significance for association with response (P=5.0E−08). Replication of this association signal is warranted, before further functional follow-up. Moreover, several of the top association signals that did not achieve genome-wide significance were in genes of biological interest, most notably NRG1, which merits further investigation into their potential role in depression and antidepressant response.
The third highest association signal in our meta-analysis of response locates to the promoter region of the neurotrophic factor neuregulin-1 (NRG1) gene. Neuregulin is involved in many aspects of brain development, including neuronal maturation, and variations in NRG1 are associated with risk for mental disorders including schizophrenia.22, 23 In fact, the SNP with strongest evidence of association with SSRI treatment outcomes identified here (rs10954808) is in linkage disequilibrium with rs7014762 (r2=0.80 in 1000 genomes CEU sample; r2=0.47 in 1000 genomes CHB/JPT sample), which was previously shown to be associated with a bipolar phenotype characterized by excellent recovery between episodes and no mood incongruent features.24 Another SNP in relatively high linkage disequilibrium with rs10954808 (rs4281084, r2=0.85 in 1000 genomes CEU sample, r2=0.61 in 1000 genomes CHB/JPT sample) was reported to be associated with transition to psychosis in at-risk individuals.25
Although NRG1 has been implicated in various psychiatric traits, this is, to the best of our knowledge, the first human pharmacogenomics study suggesting it may have a role in antidepressant treatment response. NRG1-ErbB4 signaling has a key role in the modulation of synaptic plasticity through regulating neurotransmission, and neuregulin was suggested to be a biomarker of MDD.26 In a recent animal model study, subchronic peripheral neuregulin-1beta administration increased ventral hippocampal neurogenesis and induced antidepressant-like behavior.27 Another recent study demonstrated that downregulation of NRG1-ErbB4 signaling in parvalbumin interneurons in the rat brain may contribute to the antidepressant properties of ketamine.28 In this study, pretreatment with NRG1 abolished both ketamine's antidepressant effects and ketamine-induced reduction in p-ErbB4, parvalbumin, 67-kDA isoform of glutamic acid decarboxylase (GAD67) and gamma-aminobutyric acid levels and increase in glutamate levels. Our results indicate that genetic variation in NRG1 may also influence the antidepressant effects of SSRIs.
Other SSRI-response pharmacogenomics candidate genes arising from our study, including MCPH1, RYR3, STK39, CTNNA3 and PPEF2, should be further investigated. Mutations in the human microcephalin gene MCPH1 cause the autosomal recessive disorder primary microcephaly, characterized by a congenital reduction of brain size particularly in the cerebral cortex.29 A reduction in brain size could result from increased cell death or alternatively from a diminished self-renewal potential of neuroprogenitors. A recent report provided evidence that MCPH1 controls neuroprogenitor entry into mitosis.30 Neuroprogenitor cell proliferation is an early event of neurogenesis, a process by which new neurons are continuously generated and incorporated into the nervous system. Recent research has suggested that hippocampal neurogenesis may have a key role in antidepressant action.31, 32 A recent genetic study in a Japanese population found that MCPH1 genetic variation is associated with automatic thoughts (may be risk factors for depression and anxiety) evaluated by the Depression and Anxiety Cognition Scale, a Japanese psychological questionnaire.33 MCPH1 has also been implicated in autism and schizophrenia.34, 35
Ryanodine receptor 3, a protein that in humans is encoded by the RYR3 gene, is normally enriched in hippocampal area CA1,36 suggesting a specialized role of this receptor in this area critical for depression and antidepressant action. Ryanodine receptors (RyR) increase activity-dependent calcium influx via calcium-induced calcium release. Calcium signals activate postsynaptic pathways in hippocampal neurons that underlie synaptic plasticity, learning and memory. Studies suggest that RyR2 and RyR3 isoforms have key roles in these processes.37, 38
The STK39 gene encodes a serine/threonine kinase. The protein is localized to both the cytoplasm and the nucleus, may act as a mediator of cell stress response39 and has been implicated in autism.40 A prior GWAS suggested that genetic variation in CTNNA3 (catenin alpha 3) is associated with antidepressant treatment-emergent suicidal ideation.41 Variants in CTNNA3 have also been suggested to have a role in autism and schizophrenia.31, 42 Finally, PPEF2, a calmodulin-binding protein phosphatase, has been shown to influence mGluR5 levels and prior evidence suggests it may be involved in schizophrenia.43
We investigated the association of SSRI response and %ΔHRSD with SNPs with strongest evidence for association with treatment outcomes in the two recent large collaborative pharmacogenomics studies of antidepressants.12, 13 Consistent with earlier studies, which have shown difficulties with replication of pharmacogenomic predictors of antidepressant treatment outcomes, our analyses did not replicate the prior results, with the exception of nominal evidence of association for SNP rs11624702 in MDGA2, and rs2546057 in KCTD15. In particular, our analyses provided nominally significant evidence of association of SNP rs11624702 in MDGA2 with response in the combined ISPC-AMPS-STAR*D meta-analysis. Previously, suggestive evidence of association of this SNP with antidepressant treatment outcomes was demonstrated in the genome-wide analysis of the NEWMEDS consortium sample (P=4.08 × 10−6).13 Other studies have also implicated this gene in depression-related traits and response to antidepressants. In particular, a genome-wide association study of SSRI/SNRI-induced sexual dysfunction in a Japanese cohort identified 11 MDGA2 SNPs that were significantly associated with the outcome at a genome-wide significant level.44 Furthermore, MDGA2 was identified as a potential neuroticism-related gene in a genome-wide association study,45 a finding that was subsequently replicated and extended by demonstrating association with a fatigability and asthenia subscale of harm avoidance.46 Thus, further investigation of the role of MDGA2 in depression and SSRI-treatment response is warranted.
Although this study represents one of the largest pharmacogenomics studies of antidepressant response to date, we recognize it also has certain limitations. Because a variety of SSRIs were used in the studies contributed to the consortium, this sample provides the opportunity to identify genes that contribute to the pharmacodynamic, but not pharmacokinetic, effects of these medications. Another limitation of the presented analysis is the short time period of follow-up, as generally, 6–8 weeks is considered necessary for investigating full response. However, a 4-week period is sufficient for detecting a change in most patients, and therefore analyses of the 4-week outcomes were focused solely on relative symptom change and response.
As in all studies of patients with major depression, heterogeneity between individual patients likely limits the power of genetic analyses. Differences in samples across sites, including differences in the patient populations and treatments, further increase this phenotypic heterogeneity. Thus, the overall patient sample is likely to include subgroups with different susceptibility factors for clinical depression. These might involve endogenous, neurobiological factors, temporal stress factors and other environmental factors, such as those of cultural or dietary origin. Investigation of more homogeneous patient subsamples, such as severe depression or depression with melancholic or predominantly anxious features, may lead to additional insights despite reduced sample sizes. The fact that this study included a large Asian sample is a strength, as we believe there are no prior large pharmacogenomics studies of antidepressants in Asian populations. However, ethnic heterogeneity of the sample may also have reduced power of our GWA analyses, particularly due to differences in SNP allele frequencies. The possibility that in different ethnic populations different SNPs in the same genes may have functional impact on clinical phenotypes, implies that gene-level and pathway-level, rather than SNP-level, analyses may be more powerful in racially diverse samples. Such analyses, as well as an in-depth investigation of potential differences in genomic treatment outcome predictors across racial groups, will be the focus of subsequent publications.
The lack of replicated predictors of treatment response in published pharmacogenomics studies of antidepressants has been disappointing, and has prompted questions regarding how future research in this area should proceed. Recent studies have demonstrated that very large sample sizes are needed to achieve the power to identify genetic predictors of complex psychiatric traits,47 and the same is likely to be true for antidepressant response. However, other avenues must be explored to increase power of these studies. A high degree of clinical heterogeneity in samples of treated patients, as well as the broad definition of treatment outcomes in most prior pharmacogenomics studies of antidepressant response, are likely factors contributing to the lack of significant and replicable findings. Specifically, clinical subtypes of depression may respond differently to treatment with certain medications, and medication response may be modified by different genetic variants for these clinical subtypes. Moreover, treatment outcomes in pharmacogenomic studies of MDD are usually limited to overall improvement in depression severity as measured by scales such as the HRSD. However, specific genes may impact particular aspects of drug response that contribute to the total HRSD score (for example, certain types of mood changes, changes in sleep or appetite/weight and so on). Studies that focus on more narrowly defined groups of patients, or investigate specific aspects of treatment response, may thus provide additional insights into genetic predictors of response to antidepressants.
In conclusion, the present findings provide little evidence of specific genetic factors that would markedly affect the clinical response to SSRI treatment in major depression. Also, similar to prior efforts, the results of our meta-analyses showed a lack of robust treatment outcome predictors. Nevertheless, novel biological targets for further investigation have been identified. In particular, some of the associations found (such as NRG1 or MCPH1) might reflect the important role of neuronal renewal among the mechanisms of antidepressant response.48 There is a need to replicate the top association signals with larger samples. There is also a need for closer exploration of genes showing the most marked associations in more refined and homogeneous patient subsamples, such as severe depression or depression with melancholic or predominantly anxious features.
References
Alonso J, Angermeyer MC, Bernert S, Bruffaerts R, Brugha TS, Bryson H et al. Prevalence of mental disorders in Europe: results from the European Study of the Epidemiology of Mental Disorders (ESEMeD) project. Acta Psychiatr Scand Suppl 2004; 109 (Suppl 420): 21–27.
Anderson HD, Pace WD, Libby AM, West DR, Valuck RJ . Rates of 5 common antidepressant side effects among new adult and adolescent cases of depression: a retrospective US claims study. Clin Ther 2012; 34: 113–123.
Smith AJ, Sketris I, Cooke C, Gardner D, Kisely S, Tett SE . A comparison of antidepressant use in Nova Scotia, Canada and Australia. Pharmacoepidemiol Drug Saf 2008; 17: 697–706.
Fabbri C, Di Girolamo G, Serretti A . Pharmacogenetics of antidepressant drugs: an update after almost 20 years of research. Am J Med Genet B Neuropsychiatr Genet 2013; 162B: 487–520.
Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD et al. A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry 2010; 67: 133–138.
Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry 2009; 66: 966–975.
Ji Y, Biernacka JM, Hebbring S, Chai Y, Jenkins GD, Batzler A et al. Pharmacogenomics of selective serotonin reuptake inhibitor treatment for major depressive disorder: genome-wide associations and functional genomics. Pharmacogenomics J 2013; 13: 456–463.
Laje G, McMahon FJ . Genome-wide association studies of antidepressant outcome: A brief review. Prog Neuropsychopharmacol Biol Psychiatry 2011; 35: 1553–1557.
Uher R, Perroud N, Ng MY, Hauser J, Henigsberg N, Maier W et al. Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. Am J Psychiatry 2010; 167: 555–564.
Yang J, Lee SH, Goddard ME, Visscher PM . GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Tansey KE, Guipponi M, Hu X, Domenici E, Lewis G, Malafosse A et al. Contribution of common genetic variants to antidepressant response. Biol Psychiatry 2013; 73: 679–682.
Uher R, GENDEP Investigators, MARS Investigators, STAR*D Investigators. Common genetic variation and antidepressant efficacy in major depressive disorder: a meta-analysis of three genome-wide pharmacogenetic studies. Am J Psychiatry 2013; 170: 207–217.
Tansey KE, Guipponi M, Perroud N, Bondolfi G, Domenici E, Evans D et al. Genetic predictors of response to serotonergic and noradrenergic antidepressants in major depressive disorder: a genome-wide analysis of individual-level data and a meta-analysis. PLoS Med 2012; 9: e1001326.
Fava M, Rush AJ, Trivedi MH, Nierenberg AA, Thase ME, Sackeim HA et al. Background and rationale for the sequenced treatment alternatives to relieve depression (STAR*D) study. Psychiatr Clin North Am 2003; 26: 457–494, x.
Mrazek DA, Biernacka JM, McAlpine DE, Benitez J, Karpyak VM, Williams MD et al. Treatment outcomes of depression: the pharmacogenomic research network antidepressant medication pharmacogenomic study. J Clin Psychopharmacol 2014; 34: 313–317.
Kudlow PA, Cha DS, McIntyre RS . Predicting treatment response in major depressive disorder: the impact of early symptomatic improvement. Can J Psychiatry 2012; 57: 782–788.
Stassen H, Anghelescu IG, Angst J, Boker H, Lotscher K, Rujescu D et al. Predicting response to psychopharmacological treatment: survey of recent results. Pharmacopsychiatry 2011; 44: 263–272.
Szegedi A, Jansen WT, van Willigenburg AP, van der Meulen E, Stassen HH, Thase ME . Early improvement in the first 2 weeks as a predictor of treatment outcome in patients with major depressive disorder: a meta-analysis including 6562 patients. J Clin Psychiatry 2009; 70: 344–353.
Pritchard JK, Stephens M, Donnelly P . Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Browning BL, Browning SR . A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 2009; 84: 210–223.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Li D, Collier DA, He L . Meta-analysis shows strong positive association of the neuregulin 1 (NRG1) gene with schizophrenia. Hum Mol Genet 2006; 15: 1995–2002.
Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 2002; 71: 877–892.
Georgieva L, Dimitrova A, Ivanov D, Nikolov I, Williams NM, Grozeva D et al. Support for neuregulin 1 as a susceptibility gene for bipolar disorder and schizophrenia. Biol Psychiatry 2008; 64: 419–427.
Bousman CA, Yung AR, Pantelis C, Ellis JA, Chavez RA, Nelson B et al. Effects of NRG1 and DAOA genetic variation on transition to psychosis in individuals at ultra-high risk for psychosis. Transl Psychiatry 2013; 3: e251.
Belzeaux R, Formisano-Treziny C, Loundou A, Boyer L, Gabert J, Samuelian JC et al. Clinical variations modulate patterns of gene expression and define blood biomarkers in major depression. J Psychiatr Res 2010; 44: 1205–1213.
Mahar I, Tan S, Davoli MA, Dominguez-Lopez S, Qiang C, Rachalski A et al. Subchronic peripheral neuregulin-1 increases ventral hippocampal neurogenesis and induces antidepressant-like effects. PLoS One 2011; 6: e26610.
Wang N, Zhang GF, Liu XY, Sun HL, Wang XM, Qiu LL et al. Downregulation of neuregulin 1-ErbB4 signaling in parvalbumin interneurons in the rat brain may contribute to the antidepressant properties of ketamine. J Mol Neurosci 2014; 54: 211–218.
Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, Mannan J et al. Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet 1998; 63: 541–546.
Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ . MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat Cell Biol 2011; 13: 1325–1334.
Boldrini M, Arango V . Antidepressants, age, and neuroprogenitors. Neuropsychopharmacology 2010; 35: 351–352.
Tang SW, Helmeste D, Leonard B . Is neurogenesis relevant in depression and in the mechanism of antidepressant drug action? A critical review. World J Biol Psychiatry 2012; 13: 402–412.
Ishitobi Y, Inoue A, Aizawa S, Masuda K, Ando T, Kawano A et al. Association of microcephalin 1, syntrophin-beta 1, and other genes with automatic thoughts in the Japanese population. Am J Med Genet B Neuropsychiatr Genet 2014; 165: 492–501.
Oldmeadow C, Mossman D, Evans TJ, Holliday EG, Tooney PA, Cairns MJ et al. Combined analysis of exon splicing and genome wide polymorphism data predict schizophrenia risk loci. J Psychiatr Res 2014; 52: 44–49.
Ozgen HM, van Daalen E, Bolton PF, Maloney VK, Huang S, Cresswell L et al. Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin Genet 2009; 76: 348–356.
Murayama T, Ogawa Y . Properties of Ryr3 ryanodine receptor isoform in mammalian brain. J Biol Chem 1996; 271: 5079–5084.
Adasme T, Haeger P, Paula-Lima AC, Espinoza I, Casas-Alarcon MM, Carrasco MA et al. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc Natl Acad Sci USA 2011; 108: 3029–3034.
Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G et al. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron 1999; 24: 701–713.
Balatoni CE, Dawson DW, Suh J, Sherman MH, Sanders G, Hong JS et al. Epigenetic silencing of Stk39 in B-cell lymphoma inhibits apoptosis from genotoxic stress. Am J Pathol 2009; 175: 1653–1661.
Ramoz N, Cai G, Reichert JG, Silverman JM, Buxbaum JD . An analysis of candidate autism loci on chromosome 2q24-q33: evidence for association to the STK39 gene. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 1152–1158.
Menke A, Domschke K, Czamara D, Klengel T, Hennings J, Lucae S et al. Genome-wide association study of antidepressant treatment-emergent suicidal ideation. Neuropsychopharmacology 2012; 37: 797–807.
Bacchelli E, Ceroni F, Pinto D, Lomartire S, Giannandrea M, D'Adamo P et al. A CTNNA3 compound heterozygous deletion implicates a role for alphaT-catenin in susceptibility to autism spectrum disorder. J Neurodev Disord 2014; 6: 17.
Timms AE, Dorschner MO, Wechsler J, Choi KY, Kirkwood R, Girirajan S et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry 2013; 70: 582–590.
Kurose K, Hiratsuka K, Ishiwata K, Nishikawa J, Nonen S, Azuma J et al. Genome-wide association study of SSRI/SNRI-induced sexual dysfunction in a Japanese cohort with major depression. Psychiatry Res 2012; 198: 424–429.
van den Oord EJ, Kuo PH, Hartmann AM, Webb BT, Moller HJ, Hettema JM et al. Genomewide association analysis followed by a replication study implicates a novel candidate gene for neuroticism. Arch Gen Psychiatry 2008; 65: 1062–1071.
Heck A, Pfister H, Czamara D, Muller-Myhsok B, Putz B, Lucae S et al. Evidence for associations between MDGA2 polymorphisms and harm avoidance: replication and extension of a genome-wide association finding. Psychiatr Genet 2011; 21: 257–260.
Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–994.
Gueorguieva R, Mallinckrodt C, Krystal JH . Trajectories of depression severity in clinical trials of duloxetine: insights into antidepressant and placebo responses. Arch Gen Psychiatry 2011; 68: 1227–1237.
Acknowledgements
The complete data set of clinical variables is available to registered PharmGKB users at http://www.pharmgkb.org/downloads/. The GWAS data will be made available in dbGaP. We note with sadness the passing of Dr David Mrazek before he could see this manuscript published. We honor his memory by continuing the research he inspired. We are grateful to all patients for their participation. We thank the physicians and other hospital staff, scientists, research assistants and study staff who contributed to the patient recruitment, data collection and sample preparation. We thank Professor Keh-Ming Lin for initiation of the antidepressant pharmacogenomics study and the many physicians who identified the patients and obtained the samples. This research was supported by the National Institute of Health grants U19 GM61388 (The Pharmacogenomics Research Network) and R24 GM61374, NIH/NCRR/NCATS CTSA grant number UL1 RR024150, Thailand Research Fund (TRF), Thailand Center of Excellence for Life Sciences (TCELS), National Center for Genetic Engineering and Biotechnology (BIOTEC), Tampere University Hospital Research Fund, the German Ministry of research and education in the German lead project on pharmacogenetic diagnostics grants in 2000-2004, grants from SENSHIN Medical Research Foundation and Grant-in-Aid for Scientific Research (KAKENHI), grants from the National Research Program for Genomic Medicine (NSC 98-3112-B-400-011, NSC 99-3112-B-400-003 and NSC 100-3112-B-400-015), the National Science Council (NSC 97-2314-B-400-001-MY3 and NSC 100-2314-B-400-002-MY3), the National Health Research Institutes, Taiwan (MD-095-PP-01, MD-095-PP-02, MD-097-PP-14, PH-098-PP-41, PH-098-PP-46, PH-098-PP-38, PH-098, 99-PP-42, PH-100-PP-37) and the Taiwan Psychiatric Research Network (PH-98, 99, 100-SP-11). Genotyping was funded by the RIKEN Center for Integrative Medical Science, Yokohama, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
VA is a member of the advisory boards (past 5 years) for Astra-Zeneca, Eli Lilly, Lundbeck, Otsuka, Servier, Trommsdorff. BTB is a member of advisory board (past 5 years) of Lundbeck. RW and LW own stock in OneOme LLC. RBA and TEK are stockholders in Personalis. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Biernacka, J., Sangkuhl, K., Jenkins, G. et al. The International SSRI Pharmacogenomics Consortium (ISPC): a genome-wide association study of antidepressant treatment response. Transl Psychiatry 5, e553 (2015). https://doi.org/10.1038/tp.2015.47
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.47
This article is cited by
-
Investigating genetic variants for treatment response to selective serotonin reuptake inhibitors in syndromal factors and side effects among patients with depression in Taiwanese Han population
The Pharmacogenomics Journal (2023)
-
Predicting treatment outcome in depression: an introduction into current concepts and challenges
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Barriers to genetic testing in clinical psychiatry and ways to overcome them: from clinicians’ attitudes to sociocultural differences between patients across the globe
Translational Psychiatry (2022)
-
Prediction of short-term antidepressant response using probabilistic graphical models with replication across multiple drugs and treatment settings
Neuropsychopharmacology (2021)
-
Genetic variants associated with cardiometabolic abnormalities during treatment with selective serotonin reuptake inhibitors: a genome-wide association study
The Pharmacogenomics Journal (2021)
