
Abstract
This study is, to the best of our knowledge, the first application of whole transcriptome sequencing (RNA-seq) to cells isolated from postmortem human brain by laser capture microdissection. We investigated the transcriptome of dentate gyrus (DG) granule cells in postmortem human hippocampus in 79 subjects with mental illness (schizophrenia, bipolar disorder, major depression) and nonpsychiatric controls. We show that the choice of normalization approach for analysis of RNA-seq data had a strong effect on results; under our experimental conditions a nonstandard normalization method gave superior results. We found evidence of disrupted signaling by miR-182 in mental illness. This was confirmed using a novel method of leveraging microRNA genetic variant information to indicate active targeting. In healthy subjects and those with bipolar disorder, carriers of a high- vs those with a low-expressing genotype of miR-182 had different levels of miR-182 target gene expression, indicating an active role of miR-182 in shaping the DG transcriptome for those subject groups. By contrast, comparing the transcriptome between carriers of different genotypes among subjects with major depression and schizophrenia suggested a loss of DG miR-182 signaling in these conditions.
Similar content being viewed by others

Introduction
Schizophrenia, bipolar disorder and major depression are common and severely disabling psychiatric conditions with a partially genetic background.1, 2, 3, 4 Family studies have shown co-aggregation of the major psychiatric disorders, and population-based studies have indicated shared genetic susceptibility loci.5, 6, 7, 8, 9 Further evidence for common etiological factors comes from similarities in gene expression changes observed in different diseases, which have implicated deficits in neurotransmission and mitochondrial function, elevated immune response and inflammation, and downregulation of genes expressed in oligodendrocytes.10, 11, 12, 13, 14, 15, 16, 17, 18 The goal of our study was to find common etiological mechanisms for these diseases through the identification of shared transcriptome changes.
One of the brain regions most consistently implicated in mental illness is the hippocampus, a brain region involved in memory, cognition, mood regulation and stress response.19 In subjects with schizophrenia, bipolar disorder or major depression, abnormalities in hippocampus structure or function as well as a broad range of gene expression changes have been described.12,13,20,21,22,23,24,25,26,27,28 The great majority of prior studies has been done in frontal cortex, but hippocampus has been repeatedly investigated as well.29,30 However, interpreting changes in the hippocampal transcriptome is fraught with difficulty because of the different tasks performed by different hippocampal subregions. Consequently the areas CA1, CA3, and the dentate gyrus (DG) show large differences in gene expression; additional variability is introduced by functional differentiation along the long axis of the hippocampus.31, 32, 33, 34 The DG is of particular interest as it is one of only two brain regions where adult neurogenesis has been described.35 A large body of literature has linked hippocampal neurogenesis with psychiatric illness, including affective disorders and schizophrenia.36,37 We therefore chose to investigate the transcriptome of DG granule cells, isolating them from the surrounding tissue and harvesting them by laser capture microdissection (LCM).38 We believe our study is the first to combine LCM with RNA-seq.
Materials and methods
Human subjects
Postmortem human brain tissue from 79 individuals was obtained from the Stanley Medical Research Institute (SMRI) Neuropathology Consortium, the UCLA Human Brain and Spinal Fluid Resource Center, and the University of Washington (UW) Neuropathology Core Brain bank. We investigated mid-hippocampus tissue from 79 subjects. Most (n=60) subjects were from the SMRI Neuropathology Consortium, a well-described brain collection which has been extensively used in neuropsychiatric research.39 The collection consists of four groups of 15 subjects each with schizophrenia, bipolar disorder, major depression and nonpsychiatric controls. Groups are matched by gender with nine males and six females per group, and by age, ranging from 25–68 years. Ten subjects, four males and five females ranging in age from 44–91 years old, were from the UCLA Human Brain and Spinal Fluid Resource Center. Of these, two carried a diagnosis of schizophrenia, one of bipolar disorder, two had suffered from major depression and five were nonpsychiatric controls. An additional nine nonpsychiatric controls were from the UW Neuropathology Core Brain bank. These subjects, five males and four females, ranged in age from 78–91 years.
The gender distribution (41–47% female) did not vary significantly by disease group (schizophrenia, bipolar disorder, major depression and nonpsychiatric controls). The mean age of our subjects did vary significantly by group (P<10−4), however, because our nonpsychiatric control subjects were on average older at death (64±20 years) than members of the three disease groups, schizophrenia (47±17 years), bipolar disorder (42±16 years) and major depression (47±17 years). We therefore evaluated the possible confounding influence of age by comparing DG transcriptomes between the seven youngest (age 19–44, mean 39±5.7 years) and the six oldest (age 90–95, mean 92±1.9 years) members of the healthy subject group. We identified four genes whose levels of expression appeared to be influenced by age (MAGI2, RASGRF1, USP24 and NUP107). However, none of these genes were identified in any of our disease group comparisons, indicating that our results were not influenced by the age difference between psychiatric subjects and controls.
This study was approved by the Institutional Review Board of the University of Washington and conducted in accordance with ethics guidelines for the use of human subjects in research.
Laboratory methods
Fresh frozen 14 μm slide-mounted coronal cryostat sections from mid-hippocampus were stained and dehydrated using the Arcturus HistoGene LCM frozen section staining kit (Life Technologies, Grand Island, NY, USA), and following the manufacturer’s instructions. From each subject, triplicate samples of about 2000 DG granule cells each were harvested by LCM, using an Arcturus AutoPix LCM system and CapSure Macro LCM caps (Molecular Devices, Sunnyvale, CA, USA). Triplicates were processed separately during cell harvest, RNA extraction and aRNA amplification to reduce experimental noise introduced during these stages of the experiment. Harvested cells were removed from the caps and RNA extracted using PicoPure RNA isolation kits (Life Technologies). RNA was then linearly amplified over two rounds of aRNA amplification, using MessageAmp II aRNA amplification kits (Life Technologies), and following the manufacturer’s protocol. The quality and concentration of aRNA was checked by spectrophotometry, and only samples with an A260/280 ratio >1.9 were used. Equimolar amounts of triplicate aRNA samples for each of the 79 subjects were then pooled for the preparation of sequencing libraries. To evaluate different normalization/scaling methods, a separate test data set was prepared. In this data set, duplicate aRNA samples (denoted A and B) from four randomly chosen subjects (T1–T4) were used to construct a set of eight libraries.
Sequencing libraries were prepared using Total RNA Sequencing Kit (Life Technologies), following the directions for Whole Transcriptome Libraries, and analyzed with an Applied Biosystems SOLiD 4 high-throughput sequencer with an average singe-end read length of 50 base pairs (bp) (Life Technologies).
The genetic polymorphism rs76481776 of miR-182 was genotyped using a StepOnePlus Real-Time PCR System and a TaqMan Custom SNP Genotyping Assay (Life Technologies). A amount of 50 ng genomic DNA was amplified in the presence of gene-specific primers and allele-specific fluorescent probes following the manufacturer’s instructions. Genotypes were called using TaqMan Genotyper software. For quality control 10% of the samples were genotyped in duplicate, and the genotype distribution was tested for deviation from Hardy–Weinberg equilibrium, using a χ2 test.
Data analysis
Reads were mapped and counted using the Applied Biosystems software BioScope 1.2.1. Transcripts were mapped to genome build GRCh37/hg19 (February 2009 assembly), using the UCSC RefGene annotations and the BioScope default seed-and-extend approach for mapping. Reads were mapped to the whole genome file, supplemented by a transcript annotation file allowing reads to align across known splice junctions with no gap penalty. Repeat and ambiguously mapped sequences were then removed from the counts files using a UCSC RepeatMasker file. We then used the BEDTools bamToBed program to generate files of read locations.40 We only counted reads mapping to the opposite strand. As in RNA-seq, even a gene with a single count among multiple subjects will be listed as ‘expressed’ in the sequencer output, very low-expressing genes need to be excluded by a threshold criterion to limit experimental noise.41,42 We thus only considered genes to be expressing at analyzable levels if their raw counts were greater than zero in at least 95% of subjects, that is, zero in no more than three of our 79 samples. For our test data sets which were used for the evaluation of different normalization strategies, this meant that all genes without mappable reads in any one of the eight samples were excluded (since one out of eight would have amounted to 12.5% zero reads).
The average number of total mapped reads per subject was 16 357 257. The average fraction of uniquely mapped reads was 45%. For multiple transcripts at a given genomic location, for example, owing to the presence of splice variants, only one transcript with the highest number of counts was included in the analyses. A total of 15 761 of the resulting 22 075 transcripts failed our threshold criterion of expression above background; the remaining 6314 transcripts/genes were analyzed with regard to disease-specific expression profiles. Our test data set, used for the evaluation of different normalization strategies, contained 9858 transcripts. The reason that our test data set contained a third more transcripts than the analysis data set lies with our exclusion of genes expressing at background, which led to a higher number of genes being dropped from the analysis data set.
For the evaluation of different normalization approaches, we compared a panel of 17 different methods plus raw (non-normalized) data. Normalization methods differed by their use of exon vs transcript length data and different scaling approaches (Table 1; also see Supplementary Methods for a comprehensive mathematical description). Each of these 18 approaches was applied to our test data set consisting of technical replicates from four subjects (T1–T4). We employed k-means clustering in an attempt to recover the four natural clusters in which technical replicates are paired, and evaluated methods by how well they recovered the natural clusters.
To account for the fact that genes act cooperatively in biological systems, we developed a new analysis approach for comparison of transcriptome profiles which is based on the identification of genes which are, given the presence of all other genes, significantly involved in shaping a specific gene expression profile. This regression-based analysis approach, identified by the acronym SIcall (for 'significantly involved calls') is described in detail in the Supplementary Methods. To infer microRNA (miRNA) involvement from groups of significantly involved genes, we used TargetScan (Release 6.3, June 2012). Genotype by target gene expression interactions were modeled using two-way analysis of variance (ANOVA) models (see Supplementary Methods for details).
Results
Superior performance of nonstandard normalization methods in postmortem human brain
LCM combined with aRNA amplification from postmortem human brain is a powerful technique to obtain cell population specific transcriptome data, but poses technical challenges. RNA from postmortem human brain is subject to degradation owing to agonal factors and a postmortem interval between death and the preservation of tissue. This problem is compounded by LCM, during which some amount of RNA degradation inevitably occurs even with stringent RNAse-free technique.43 In addition, aRNA production leads to shortening of transcriptomes over successive cycles of amplification.44, 45, 46
In the most widely used normalization strategy for RNA-seq experiments, the RPKM method (reads per kilobase of exon model per million mapped reads), dividing raw counts by exon length reduces the bias that is introduced by the fact that longer genes accumulate more counts.47 In our sample of 96 subjects, average mapped reads (raw counts) were more strongly proportional to total transcript length (r=0.427, P<10−4) than to cumulative exon length, however (r=0.080, P<10−4) (see also the Supplementary Figure).
In principle, the shortening of measurable transcripts occurring as a result of partial mRNA degradation and aRNA amplification under our experimental condition disproportionally affects shorter genes. For example, a loss of 500 bp will remove 50% of the signal from a 1-kb transcript, but only 25% of the signal of a 2-kb mRNA. As a result, in our experiment shorter genes had ‘noisier’ levels of expression, as indicated by higher coefficients of variation of the raw mapped counts. This inversely proportional relationship was stronger for total transcript length (r=−0.104, P<10−4) than for exon length (r=−0.063, P<10−4).
To answer the question which normalization method would perform best under our experimental conditions, we designed and tested 17 different methods, plus no normalization. K-means clustering was used to compare the quality of different approaches, based on the assumption that the best performing method(s) would appropriately cluster technical replicates together and samples from the four different subjects as distinct. Only one method (#16) led to the correct clustering; two other methods produced results that were identical to each other with one subject placed in the wrong cluster (#9 and #17, Table 1). The top-performing method filters out a portion of the experimental noise by introducing a stronger bias against noisier, shorter transcripts, whereas the two runners-up reduce the inherent sequencing bias against short transcripts in a way that is similar to the RPKM method. As the top-performing method, noise reduction scaling (#16) became the basis for our subsequent analyses. Length scaling (# 9) was used to compare the effects of different normalization strategies, and to confirm results.
Identification of transcriptome differences between subject groups
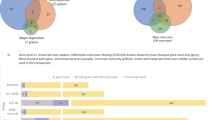
We made the following seven comparisons (1) all mental illness (n=50) vs nonpsychiatric controls (n=29), (2) schizophrenia (n=17) vs nonpsychiatric controls (n=29), (3) bipolar disorder (n=16) vs nonpsychiatric controls (n=29), (4) major depression (n=17) vs nonpsychiatric controls (n=29), (5) schizophrenia (n=17) vs bipolar disorder (n=16), (6) schizophrenia (n=17) vs major depression (n=17), (7) bipolar disorder (n=16) vs major depression (n=17). Cumulatively these comparisons identified 141 genes as likely to be involved in shaping DG expression profiles in mental illness (Supplementary Table 1). In contrast the ‘fold change’ output of traditional regression methods, the weights given by SIcall analysis are a rough measure of the likelihood of a gene contributing to overall DG gene expression changes in mental illness, given the presence of all other genes represented in the transcriptome.
Influence of the normalization and analysis method on gene identification
Using an alternative normalization method, length scaling instead of noise reduction scaling led to the identification of 162 genes. Only 64 of these genes, in particular the most heavily weighted ones, were identified using both scaling methods. Hence, identification of the majority of genes was strongly dependent on the choice of normalization method. For the 64 genes identified in both versions of the scaled data, there was fairly good overlap in the pattern of weights generated by our different comparisons (Supplementary Table 2).
The dependence of gene identification on normalization method was independent of the type of analysis method used. Using univariate logistic regression models led to the identification of 65 genes as differentially expressed (any of seven group comparisons, P<0.01) using length scaled data, and 80 genes as differentially expressed (any of seven group comparisons, P<0.01) using noise reduction scaling, with an overlap of 26 genes between the two normalization methods (data not shown). Hence, for either analysis method (SIcall vs a more traditional approach) only about a third of genes were reproducibly identified across different normalization methods.
Deducing miRNA involvement from transcriptome data
We next investigated the possibility that our observed DG transcriptome changes in mental illness could be the result of dysregulated miRNA signaling. The deduction of miRNA involvement from mRNA gene expression profiles relies on computational approaches that match miRNAs to target genes by searching the 3′UTR of potential miRNA target genes for 6–8 bp miRNA binding sites. Yet only a subset of miRNA binding sites and target genes identified by purely computational approaches is biologically relevant.
To address this problem we applied a two-step approach in which we used our most heavily weighted genes (Set 1, weights of 4 or greater, n=21) to discover possible miRNA involvement, using TargetScan. Genes with a total comparison weight 1–3 were assigned to Set 2 (n=117) (Supplementary Table 1). We hypothesized that if DG transcriptome changes in psychiatric conditions result at least to some extent from dysregulation of signaling by a miRNA, both heavily weighted (Set 1) and more lightly weighted genes (Set 2) should have overrepresentation of target genes for this particular miRNA compared with the remainder of transcripts expressing above background but not identified by SIcall (‘non-called genes’, NC, n=6055). We further hypothesized that targeting by this miRNA would be strongest in Set 1 genes, followed by Set 2 genes, followed by NC genes.
Twenty miRNAs or miRNA families targeted at least 25% (⩾5) of Set 1 genes (Supplementary Table 3). Among these, two followed the hypothesized pattern of a drop in targeting rates from Set 1 over Set 2 to NC genes, with statistically significant differences in the number of targeted genes between Set 2 and NC genes: miR-182 and the miR-30abcdef/30abe-5p/384-5p family. After Bonferroni correction for the number of χ2 tests performed, only miR-182 remained statistically significant (Table 2).
A higher proportion of miR-182 target genes among genes identified by SIcall compared with NC genes was also observed if length scaling was used for normalization. A total of 27 of 158 genes identified by SIcall (any weight) in our method 9 normalized data set were miR-182 targets, compared with 636 targets among the corresponding 6035 NC genes (χ2=6.9, P=0.009).
Validation of miR-182 involvement in shaping DG granule cell transcriptomes
Saus et al.48 have shown that a C to T substitution in the single-nucleotide polymorphism rs76481776 leads to overexpression of miR-182 in T- vs C-allele carriers and causes a significant reduction in target gene expression. The minor allele frequency of rs76481776 in our subjects was 8.9%, that is, 13 of our 79 subjects were T-allele carriers, one of them a T/T homozygote; the remaining 66 individuals had the C/C genotype. Our minor allele (T) frequency of 8.9% was in good agreement with the previously reported 7.5% in Spanish subjects.48 Genotypes were in Hardy–Weinberg equilibrium (not shown).
We hypothesized that whenever miR-182 was active in shaping DG gene expression profiles, we would be able to observe a statistically significant difference in target gene expression between T-allele carriers (C/T or T/T genotype) and those with the C/C genotype. On the other hand, no difference in miR-182 target gene expression between rs76481776 T-allele carriers vs noncarriers would indicate that miR-182 is not actively involved in regulating the transcriptome. To use the functional variant rs76481776 as a detector of miRNA-182 action on the transcriptome, we compared the expression levels of miR-182 target genes between carriers and noncarriers of the uncommon T-variant in each of our three disease groups (schizophrenia, bipolar disorder, major depression) and in nonpsychiatric controls.
The differences in the mean miR-182 target expression levels between carriers and noncarriers of the rs76481776 T-allele varied as a function of the psychiatric diagnosis (F=13.10, P<10−4). We observed significant differences in mean miR-182 target expression levels between carriers and noncarriers of the T-allele in nonpsychiatric controls (t=4.77, P<10−4) and in subjects with bipolar disorder (t=−3.48, P<10−4). By contrast, target gene expression levels did not differ significantly by genotype group in individuals with schizophrenia (t=−1.61, P=0.108) or major depression (t=0.88, P=0.380). Hence, although miR-182 targeting is active in DG granule cells of control subjects and individuals with bipolar disorder, it appears to be lost in subjects with schizophrenia and major depression. Using our alternative normalization method (length scaled data) we could confirm loss of miR-182 signaling in subjects with depression, but not in schizophrenia (data not shown).
Discussion
This study is, to the best of our knowledge, the first application of RNA-seq to cells isolated from postmortem human brain by LCM. Our whole transcriptome analysis approach, SIcall, will be a useful addition to the tool chest of other currently available analysis methods. To our knowledge, our study is also the first to use genetic methods for validation of miRNA gene targeting in global transcriptomes.
Gene expression studies in humans are made difficult by the marked heterogeneity of subjects, which strongly reduces statistical power.49 We therefore tried to maximize our power to detect differences between groups by collecting a larger subject group, using samples from three different brain banks. Nonetheless, variability of subject and sample characteristics creates important confounders in transcriptome comparisons. Previous studies have indicated that RNA quality, brain pH, postmortem interval, subject gender, ethnicity, age, disease duration, drug treatment history, suicide status, alcohol and substance abuse comorbidity can affect results.13 Among these, RNA integrity and the factors directly affecting it such as postmortem interval and brain pH have by far the strongest impact.50,51 Analysis of postmortem human brain has shown that a longer postmortem interval and lower tissue pH lead to decreased RNA integrity, which introduces noise into gene expression data.52, 53, 54 This is further exacerbated by a previously demonstrated 30% drop in RNA integrity during LCM.43 Other covariates may create false positive reports of differential gene expression or mitigate true gene expression differences. For example, a previous study of SMRI samples has shown that higher cumulative lifetime antipsychotic dose probably normalizes some of the inherent molecular changes of schizophrenia.55 Additional difficulty is created by the fact that not all relevant subject information might be known. For example we did not have access to family psychiatric history or lifetime exposure to psychotropic drugs for subjects from the UCLA and UW brain banks. We investigated the influence of subject age, our potentially most relevant confounder, on transcriptome differences, and found that it did not affect our results. Given the large number of potentially confounding subject variables, however, we cannot exclude the possibility that other confounders might have influenced our observed gene expression patterns.
The statistical analysis of transcriptome data traditionally relies on separate comparison of expression levels for each gene between case and control conditions, resulting in a report of fold changes for each gene. To explore the cooperative action of groups of genes, we used an alternative regression-based analysis approach (SIcall) looking at the simultaneous actions of up to five genes. Although models with higher numbers of participating genes are possible, their computational cost is prohibitive. For each model, our algorithm generates a large number of logistical regressions representing the many ways in which small groups of genes can cooperatively characterize transcriptome differences between two groups. Similar approaches have previously been used in the analysis of gene expression data.56 For each comparison we set the threshold of the probability at which a gene would be considered involved in shaping gene expression profiles to 0.05. In other words, if a gene had at least 5% probability of being featured in one of five sets of logistic regression models allowing for the simultaneous action of either 1, 2, 3, 4 or 5 genes at a time, it was listed as significantly involved and entered our subsequent analysis steps. It should be noted that this 5% represents an empirically chosen probability threshold which does not correspond to statistical significance. Genes were weighted by the number of times they were called per comparison (up to five), and the total weights across all seven comparisons (up to a theoretical maximum of 35). It should be noted that total gene weights are not quantitative in the way gene expression changes are, but they rather represent rough measures of the likelihood of the involvement of a given gene. Only the results of the simplest SIcall models, such as those which investigate one gene at a time, roughly correspond to the traditional idea of gene-by-gene differential expression.
Including individual disease vs disease comparisons (e.g. schizophrenia vs major depression) in our analysis was based on the hypothesis that any gene significantly involved in shaping a disease-specific transcriptome might also reveal itself in comparison of this disease with any other psychiatric condition. For example, the gene C9orf102 is heavily weighted in both the bipolar disorder vs control, and the bipolar disorder vs depression comparisons. We can hypothesize from this that C9orf102 expression might potentially be useful as a biomarker differentiating bipolar disorder from major depression, warranting further experimental exploration and confirmation. The genes OPTN, FAM124A, OXSR1, RLF and TLL1 are heavily weighted in the comparison of schizophrenia against major depression, but are not called in any other comparison. Our analysis does not reveal which of these genes might be involved in schizophrenia, which in depression, or which in both, the latter as a result of opposing gene expression changes in the two conditions. Nonetheless, the fact that they are called by our analysis indicates that investigating them further might yield insights into broad processes which may be dysregulated in major depression or schizophrenia. Our inclusion of an ‘all disease’ vs nonpsychiatric control comparison was motivated by the hypothesis that major psychiatric conditions might share subtle transcriptome changes that are detectable only if larger groups are compared. However, contrary to this expectation, there were relatively few genes that had weights >1 and were called only in the all disease vs control, but not in any other comparison.
Prior gene expression studies in postmortem human brain have successfully implicated broad systems dysfunction in mental illness.10, 11, 12, 13, 14, 15 Most of these studies have used microarray technology, but RNA-seq has been employed in more recent work.16, 17, 18 The vast majority of prior studies investigated tissue blocks as opposed to near-homogeneous cell populations isolated by LCM. One previous study exists in which DG gene expression profiles were compared in subjects with schizophrenia, bipolar disorder, major depression and nonpsychiatric controls, using LCM and microarrays.57 The authors found decreased expression of genes related to protein turnover, energy metabolism and neuronal functions in subjects with schizophrenia compared with controls. No significant transcriptome changes were observed in subjects with major depression or bipolar disorder.
Although prior gene expression studies have been consistent in reporting systems-level dysfunction in the brains of subjects with mental illness, for example, inflammation, observations of differential expression for individual genes have been far less reproducible. In a meta-analysis of 12 genome-wide expression studies in postmortem brain of subjects with bipolar disorder compared with controls, Elashoff et al.14 have shown that the likelihood of a gene reported as differentially expressed in one study having a repeat finding in one of 11 other studies was only 9%. This lack of robustness in gene expression findings has previously been attributed to interacting factors such as tissue pH and subject age or gender.13 Our findings indicate that the choice of suboptimal normalization methods may be an additional contributing factor. Under our experimental conditions the generally accepted standard of transcriptome normalization, the RPKM method, showed inferior performance compared with the other approaches.
We hypothesized that our observed gene expression changes might have occurred as the result of an overarching dysregulation of gene expression in mental illness. We chose to look at posttranscriptional regulation by miRNAs because of strong evidence of their involvement in DG neurogenesis and major psychiatric disorders. The miRNAs are a class of small noncoding RNAs which inhibit expression of groups of target genes through degradation or inhibition of their mRNAs. Over half of miRNAs are highly or exclusively expressed in brain, where they participate in neurogenesis and neuronal plasticity.58, 59, 60, 61, 62, 63, 64, 65 Changes in miRNA expression in postmortem human brain have previously been shown in schizophrenia,66, 67, 68, 69, 70, 71, 72, 73, 74 bipolar disorder72,73 and depression.75
Contrasting with our approach, prior studies have relied on the direct profiling of miRNA expression using a preselected panel of miRNAs. Accurate direct profiling of miRNAs in postmortem human brain could be compromised, however, by potentially limited miRNA stability in neuronal cells. Although studies in embryonic cell lines and peripheral organs such as liver and heart have shown that miRNAs can be highly stable molecules with half-lives of up to several days,76, 77, 78 comparing miRNA decay in neurons with that in non-neuronal cells, Krol et al.79 found miRNA decay in neurons to be activity-dependent, and occurring much faster than in non-neuronal cells. Their observation of miRNA half-lives of less than 1 h agrees with other findings in human primary neuronal cells and short postmortem interval human neocortex, where Sethi and Lukiw80 reported miRNA half-lives ranging from 1 h to about 3.5 h. The latter would mean that during a postmortem interval of 24 h, which is the case for many human subjects in publicly available brain collections, more than 99% of miRNA molecules have degraded.50 Our ability to directly detect changes in miRNA expression in our subjects was further compromised by our use of aRNA amplification. Although miRNAs are translated from polyadenylated transcripts, the poly-A tail is lost in mature miRNAs. Hence the bulk of mature miRNAs was lost during aRNA amplification before the preparation of sequencing libraries. Possibly as a result of that, we were not able to detect expression of mir-182 or any member of the miR-30 family in our subjects.
We believe our study is the first to show evidence of disrupted miR-182 signaling in schizophrenia and major depression. Members of the miR-30 family, however, have been repeatedly shown to have decreased expression in schizophrenia.81 It should be noted that only a minority of genes identified in our RNA-seq analysis were actually miR-182 targets. Hence, it is clear that we were able to discover only one of possibly a multitude of regulatory mechanisms accountable for shaping DG transcriptome changes in mental illness.
miR-182 is part of a cluster of three miRNAs, miR-96, miR-182 and miR-183, which are colocated within a 4 kb genomic segment located at 7q32.2.82,83 miR-182 is involved in a broad range of biological processes including regulation of the immune response, DNA repair, cell proliferation and differentiation, and regeneration of peripheral nerves after injury.84, 85, 86, 87, 88, 89 miR-182 is the highest expressing miRNA in the pineal gland, where it accounts for 28% of the miRNA population. There, miR-182 has a rhythmic pattern of gene expression involving approximately two-fold changes between the highest levels of expression between 6 am and 12 pm, and the lowest levels during the night.90 In keeping with this, Saus et al.48 found an association between the rs76481776 polymorphism of the miR-182 gene and patterns of insomnia in patients with major depression. Disruption of sleep and circadian rhythmicity are cardinal features of both schizophrenia and major depression, which aligns with our findings of a possible loss of miR-182 signaling in these conditions.91, 92, 93
Conclusions
Whole transcriptome analysis by RNA-seq in LCM-isolated DG granule cells of postmortem hippocampus in subjects with mental illness and controls showed evidence of disrupted miR-182 signaling in subjects with major depression and schizophrenia. We validated this finding by showing how the impact of a functional miR-182 single-nucleotide polymorphism on target gene expression was lost in subjects with schizophrenia and major depression. Our paper is the first study in which LCM is combined with RNA-seq in postmortem human brain. Under these challenging experimental conditions, a noise reduction scaling normalization method outperformed normalization by exon or transcript length. We also demonstrate the feasibility of a novel, regression-based method for RNA-seq analysis (SIcall) which allows for the investigation of cooperative action among small sets of genes and is a useful complement to existing approaches.
References
Mathers C, Fat DM, Boerma JT, World Health Organization. The Global Burden of Disease: 2004 Update. World Health Organization: Geneva, Switzerland, 2008vii, p 146.
Sullivan PF . The genetics of schizophrenia. PLoS Med 2005; 2: e212.
Sullivan PF, Neale MC, Kendler KS . Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000; 157: 1552–1562.
Smoller JW, Finn CT . Family, twin, and adoption studies of bipolar disorder. Am J Med Genet C Semin Med Genet 2003; 123C: 48–58.
Valles V, Van Os J, Guillamat R, Gutierrez B, Campillo M, Gento P et al. Increased morbid risk for schizophrenia in families of in-patients with bipolar illness. Schizophr Res 2000; 42: 83–90.
McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A . The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 2003; 60: 497–502.
Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 2009; 373: 234–239.
Blackwood DH, Pickard BJ, Thomson PA, Evans KL, Porteous DJ, Muir WJ . Are some genetic risk factors common to schizophrenia, bipolar disorder and depression? Evidence from DISC1, GRIK4 and NRG1. Neurotox Res 2007; 11: 73–83.
Blackwood DH, Fordyce A, Walker MT St, Clair DM, Porteous DJ, Muir WJ . Schizophrenia and affective disorders—cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet 2001; 69: 428–433.
Konradi C, Sillivan SE, Clay HB . Mitochondria, oligodendrocytes and inflammation in bipolar disorder: evidence from transcriptome studies points to intriguing parallels with multiple sclerosis. Neurobiol Dis 2012; 45: 37–47.
Clay HB, Sillivan S, Konradi C . Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci 2011; 29: 311–324.
Altar CA, Vawter MP, Ginsberg SD . Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology 2009; 34: 18–54.
Sequeira PA, Martin MV, Vawter MP . The first decade and beyond of transcriptional profiling in schizophrenia. Neurobiol Dis 2012; 45: 23–36.
Elashoff M, Higgs BW, Yolken RH, Knable MB, Weis S, Webster MJ et al. Meta-analysis of 12 genomic studies in bipolar disorder. J Mol Neurosci 2007; 31: 221–243.
Sokolov BP . Oligodendroglial abnormalities in schizophrenia, mood disorders and substance abuse. Comorbidity, shared traits, or molecular phenocopies? Int J Neuropsychopharmacol 2007; 10: 547–555.
Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 2013; 18: 206–214.
Hwang Y, Kim J, Shin JY, Kim JI, Seo JS, Webster MJ et al. Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Transl Psychiatry 2013; 3: e321.
Wu JQ, Wang X, Beveridge NJ, Tooney PA, Scott RJ, Carr VJ et al. Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLoS One 2012; 7: e36351.
DeCarolis NA, Eisch AJ . Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology 2010; 58: 884–893.
Adriano F, Caltagirone C, Spalletta G . Hippocampal volume reduction in first-episode and chronic schizophrenia: a review and meta-analysis. Neuroscientist 2012; 18: 180–200.
Shepherd AM, Laurens KR, Matheson SL, Carr VJ, Green MJ . Systematic meta-review and quality assessment of the structural brain alterations in schizophrenia. Neurosci Biobehav Rev 2012; 36: 1342–1356.
Frey BN, Andreazza AC, Nery FG, Martins MR, Quevedo J, Soares JC et al. The role of hippocampus in the pathophysiology of bipolar disorder. Behav Pharmacol 2007; 18: 419–430.
Chen CH, Suckling J, Lennox BR, Ooi C, Bullmore ET . A quantitative meta-analysis of fMRI studies in bipolar disorder. Bipolar Disord 2011; 13: 1–15.
Eker C, Gonul AS . Volumetric MRI studies of the hippocampus in major depressive disorder: Meanings of inconsistency and directions for future research. World J Biol Psychiatry 2010; 11: 19–35.
McKinnon MC, Yucel K, Nazarov A, MacQueen GM . A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci 2009; 34: 41–54.
Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res 2011; 131: 165–173.
Konradi C, Zimmerman EI, Yang CK, Lohmann KM, Gresch P, Pantazopoulos H et al. Hippocampal interneurons in bipolar disorder. Arch Gen Psychiatry 2011; 68: 340–350.
Walton NM, Zhou Y, Kogan JH, Shin R, Webster M, Gross AK et al. Detection of an immature dentate gyrus feature in human schizophrenia/bipolar patients. Transl Psychiatry 2012; 2: e135.
Higgs BW, Elashoff M, Richman S, Barci B . An online database for brain disease research. BMC Genomics 2006; 7: 70.
Kim S, Webster MJ . The stanley neuropathology consortium integrative database: a novel, web-based tool for exploring neuropathological markers in psychiatric disorders and the biological processes associated with abnormalities of those markers. Neuropsychopharmacology 2010; 35: 473–482.
Datson NA, Meijer L, Steenbergen PJ, Morsink MC, van der Laan S, Meijer OC et al. Expression profiling in laser-microdissected hippocampal subregions in rat brain reveals large subregion-specific differences in expression. Eur J Neurosci 2004; 20: 2541–2554.
Lein ES, Zhao X, Gage FH . Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci 2004; 24: 3879–3889.
Thompson CL, Pathak SD, Jeromin A, Ng LL, MacPherson CR, Mortrud MT et al. Genomic anatomy of the hippocampus. Neuron 2008; 60: 1010–1021.
Christensen T, Bisgaard CF, Nielsen HB, Wiborg O . Transcriptome differentiation along the dorso-ventral axis in laser-captured microdissected rat hippocampal granular cell layer. Neuroscience 2010; 170: 731–741.
Hanson ND, Owens MJ, Nemeroff CB . Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology 2011; 36: 2589–2602.
Balu DT, Lucki I . Adult hippocampal neurogenesis: regulation, functional implications, and contribution to disease pathology. Neurosci Biobehav Rev 2009; 33: 232–252.
Samuels BA, Hen R . Neurogenesis and affective disorders. Eur J Neurosci 2011; 33: 1152–1159.
Cheng L, Zhang S, Maclennan GT, Williamson SR, Davidson DD, Wang M et al. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications. Appl Immunohistochem Mol Morphol 2012; 21: 31–47.
Torrey EF, Webster M, Knable M, Johnston N, Yolken RH . The stanley foundation brain collection and neuropathology consortium. Schizophr Res 2000; 44: 151–155.
Quinlan AR, Hall IM . BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26: 841–842.
Ueda HR, Hayashi S, Matsuyama S, Yomo T, Hashimoto S, Kay SA et al. Universality and flexibility in gene expression from bacteria to human. Proc Natl Acad Sci USA 2004; 101: 3765–3769.
Bullard JH, Purdom E, Hansen KD, Dudoit S . Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 2010; 11: 94.
Wang S, Wang L, Zhu T, Gao X, Li J, Wu Y et al. Improvement of tissue preparation for laser capture microdissection: application for cell type-specific miRNA expression profiling in colorectal tumors. BMC Genomics 2010; 11: 163.
Li Y, Li T, Liu S, Qiu M, Han Z, Jiang Z et al. Systematic comparison of the fidelity of aRNA, mRNA and T-RNA on gene expression profiling using cDNA microarray. J Biotechnol 2004; 107: 19–28.
Zhu B, Xu F, Baba Y . An evaluation of linear RNA amplification in cDNA microarray gene expression analysis. Mol Genet Metab 2006; 87: 71–79.
Clement-Ziza M, Gentien D, Lyonnet S, Thiery JP, Besmond C, Decraene C . Evaluation of methods for amplification of picogram amounts of total RNA for whole genome expression profiling. BMC Genomics 2009; 10: 246.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B . Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5: 621–628.
Saus E, Soria V, Escaramis G, Vivarelli F, Crespo JM, Kagerbauer B et al. Genetic variants and abnormal processing of pre-miR-182, a circadian clock modulator, in major depression patients with late insomnia. Hum Mol Genet 2010; 19: 4017–4025.
Wei C, Li J, Bumgarner RE . Sample size for detecting differentially expressed genes in microarray experiments. BMC Genomics 2004; 5: 87.
Weis S, Llenos IC, Dulay JR, Elashoff M, Martinez-Murillo F, Miller CL . Quality control for microarray analysis of human brain samples: the impact of postmortem factors, RNA characteristics, and histopathology. J Neurosci Methods 2007; 165: 198–209.
Vawter MP, Tomita H, Meng F, Bolstad B, Li J, Evans S et al. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry 2006; 11 7: 663–679.
Webster MJ . Tissue preparation and banking. Prog Brain Res 2006; 158: 3–14.
Chevyreva I, Faull RL, Green CR, Nicholson LF . Assessing RNA quality in postmortem human brain tissue. Exp Mol Pathol 2008; 84: 71–77.
Birdsill AC, Walker DG, Lue L, Sue LI, Beach TG . Postmortem interval effect on RNA and gene expression in human brain tissue. Cell Tissue Bank 2011; 12: 311–318.
Chan MK, Tsang TM, Harris LW, Guest PC, Holmes E, Bahn S . Evidence for disease and antipsychotic medication effects in post-mortem brain from schizophrenia patients. Mol Psychiatry 2011; 16: 1189–1202.
Rich JN, Hans C, Jones B, Iversen ES, McLendon RE, Rasheed BK et al. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res 2005; 65: 4051–4058.
Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y et al. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry 2005; 58: 85–96.
Mellios N, Sugihara H, Castro J, Banerjee A, Le C, Kumar A et al. miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat Neurosci 2011; 14: 1240–1242.
Dajas-Bailador F, Bonev B, Garcez P, Stanley P, Guillemot F, Papalopulu N . microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nature Neuroscience advance online publication, 8 April 2012; doi:10.1038/nn.3082.
Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci USA 2010; 107: 20382–20387.
Lambert TJ, Storm DR, Sullivan JM . MicroRNA132 modulates short-term synaptic plasticity but not basal release probability in hippocampal neurons. PLoS One 2010; 5: e15182.
Luikart BW, Perederiy JV, Westbrook GL . Dentate gyrus neurogenesis, integration and microRNAs. Behav Brain Res 2012; 227: 348–355.
Xu B, Karayiorgou M, Gogos JA . MicroRNAs in psychiatric and neurodevelopmental disorders. Brain Res 2010; 1338: 78–88.
Miller BH, Wahlestedt C . MicroRNA dysregulation in psychiatric disease. Brain Res 2010; 1338: 89–99.
Im HI, Kenny PJ . MicroRNAs in neuronal function and dysfunction. Trends Neurosci 2012; 35: 325–334.
Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA et al. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol 2007; 8: R27.
Beveridge NJ, Tooney PA, Carroll AP, Gardiner E, Bowden N, Scott RJ et al. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet 2008; 17: 1156–1168.
Zhu Y, Kalbfleisch T, Brennan MD, Li Y . A MicroRNA gene is hosted in an intron of a schizophrenia-susceptibility gene. Schizophr Res 2009; 109: 86–89.
Beveridge NJ, Gardiner E, Carroll AP, Tooney PA, Cairns MJ . Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol Psychiatry 2010; 15: 1176–1189.
Santarelli DM, Beveridge NJ, Tooney PA, Cairns MJ . Upregulation of dicer and microRNA expression in the dorsolateral prefrontal cortex Brodmann area 46 in schizophrenia. Biol Psychiatry 2011; 69: 180–187.
Mellios N, Galdzicka M, Ginns E, Baker SP, Rogaev E, Xu J et al. Gender-specific reduction of estrogen-sensitive small RNA, miR-30b, in subjects with schizophrenia. Schizophr Bull 2012; 38: 433–443.
Kim AH, Reimers M, Maher B, Williamson V, McMichael O, McClay JL et al. MicroRNA expression profiling in the prefrontal cortex of individuals affected with schizophrenia and bipolar disorders. Schizophr Res 2010; 124: 183–191.
Moreau MP, Bruse SE, David-Rus R, Buyske S, Brzustowicz LM . Altered microRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol Psychiatry 2011; 69: 188–193.
Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci USA 2012; 109: 3125–3130.
Smalheiser NR, Lugli G, Rizavi HS, Torvik VI, Turecki G, Dwivedi Y . MicroRNA expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PLoS One 2012; 7: e33201.
Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D et al. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 2011; 39: 5692–5703.
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN . Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007; 316: 575–579.
Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 2009; 23: 1313–1326.
Krol J, Busskamp V, Markiewicz I, Stadler MB, Ribi S, Richter J et al. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 2010; 141: 618–631.
Sethi P, Lukiw WJ . Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer's disease temporal lobe neocortex. Neurosci Lett 2009; 459: 100–104.
Beveridge NJ, Cairns MJ . MicroRNA dysregulation in schizophrenia. Neurobiol Dis 2012; 46: 263–271.
Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 2005; 33: 2697–2706.
Mihelich BL, Khramtsova EA, Arva N, Vaishnav A, Johnson DN, Giangreco AA et al. miR-183-96-182 cluster is overexpressed in prostate tissue and regulates zinc homeostasis in prostate cells. J Biol Chem 2011; 286: 44503–44511.
Ceribelli A, Satoh M, Chan EK . MicroRNAs and autoimmunity. Curr Opin Immunol 2012; 24: 686–691.
Yao E, Ventura A . A new role for miR-182 in DNA repair. Mol Cell 2011; 41: 135–137.
Wang XR, Zhang XM, Du J, Jiang H . MicroRNA-182 regulates otocyst-derived cell differentiation and targets T-box1 gene. Hear Res 2012; 286: 55–63.
Kim KM, Park SJ, Jung SH, Kim EJ, Jogeswar G, Ajita J et al. miR-182 is a negative regulator of osteoblast proliferation, differentiation, and skeletogenesis through targeting FoxO1. J Bone Miner Res 2012; 27: 1669–1679.
Yu B, Qian T, Wang Y, Zhou S, Ding G, Ding F et al. miR-182 inhibits Schwann cell proliferation and migration by targeting FGF9 and NTM, respectively at an early stage following sciatic nerve injury. Nucleic Acids Res 2012; 40: 10356–10365.
Aldrich BT, Frakes EP, Kasuya J, Hammond DL, Kitamoto T . Changes in expression of sensory organ-specific microRNAs in rat dorsal root ganglia in association with mechanical hypersensitivity induced by spinal nerve ligation. Neuroscience 2009; 164: 711–723.
Clokie SJ, Lau P, Kim HH, Coon SL, Klein DC . MicroRNAs in the pineal gland: miR-483 regulates melatonin synthesis by targeting arylalkylamine n-acetyltransferase. J Biol Chem 2012; 287: 25312–25324.
Pritchett D, Wulff K, Oliver PL, Bannerman DM, Davies KE, Harrison PJ et al. Evaluating the links between schizophrenia and sleep and circadian rhythm disruption. J Neural Transm 2012; 119: 1061–1075.
Wulff K, Dijk DJ, Middleton B, Foster RG, Joyce EM . Sleep and circadian rhythm disruption in schizophrenia. Br J Psychiatry 2012; 200: 308–316.
Kronfeld-Schor N, Einat H . Circadian rhythms and depression: human psychopathology and animal models. Neuropharmacology 2012; 62: 101–114.
Acknowledgements
Postmortem brain tissue was donated by The Stanley Medical Research Institute's brain collection, the Human Brain and Spinal Fluid Resource Center and the University of Washington Neuropathology Core Brain Bank. We are very grateful to Dr Rajinder Kaul and Mr Daniel Bates at the Seattle High Throughput Genomics Center for advice and technical assistance with sequencing. We thank Ms Sarah Symonds for technical assistance. This work was supported by NIH/NIMH grant K08MH076098.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Kohen, R., Dobra, A., Tracy, J. et al. Transcriptome profiling of human hippocampus dentate gyrus granule cells in mental illness. Transl Psychiatry 4, e366 (2014). https://doi.org/10.1038/tp.2014.9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.9
Keywords
This article is cited by
-
The role of microRNAs in diseases and related signaling pathways
Molecular Biology Reports (2022)
-
Programmed cell death 4 as an endogenous suppressor of BDNF translation is involved in stress-induced depression
Molecular Psychiatry (2021)
-
Hippocampal subfield transcriptome analysis in schizophrenia psychosis
Molecular Psychiatry (2021)
-
Estrogen receptor α drives pro-resilient transcription in mouse models of depression
Nature Communications (2018)
-
Post-mortem molecular profiling of three psychiatric disorders
Genome Medicine (2017)
