
Abstract
The serotonin transporter gene-linked polymorphic region (5-HTTLPR) has been implicated in moderating vulnerability to stress-related psychopathology upon exposure to environmental adversity. A recent meta-analysis suggests a potential biological pathway conveying genotype-dependent stress sensitivity by demonstrating a small, but significant association of 5-HTTLPR and cortisol stress reactivity. An arguably more potent approach to detect larger effects when investigating the 5-HTTLPR stress sensitivity hypothesis is to account for both genetic and epigenetic variation in the serotonin transporter gene (SLC6A4). Here, we applied this approach in an experimental setting. Two hundred healthy adults were exposed to a laboratory stressor (Trier Social Stress Test) and cortisol response patterns were assessed as a function of 5-HTTLPR and DNA methylation profiles in SLC6A4. Specifically, we analyzed 83 CpG sites within a 799-bp promoter-associated CpG island of SLC6A4 using a highly sensitive bisulfite pyrosequencing method. Our results suggest that SLC6A4 methylation levels significantly moderate the association of 5-HTTLPR and cortisol stress reactivity. For individuals displaying low levels of SLC6A4 methylation, the S allele relates to increased cortisol stress reactivity in a dose-dependent fashion accounting for 7–9% of the variance in the endocrine stress response. By contrast, no such effect occurred under conditions of high SLC6A4 methylation, indicating that epigenetic changes may compensate for genotype-dependent differences in stress sensitivity. Studying epigenetic markers may advance gene–environment interaction research on 5-HTTLPR as they possibly capture the net effects of environmental influences relevant for stress-related phenotypes under serotonergic control.
Similar content being viewed by others

Introduction
Altered regulation of the hypothalamus–pituitary adrenal (HPA) axis, one of the body’s major stress systems, has been implicated as a correlate or even causal factor for a broad range of stress-related psychiatric disorders.1 While it is widely recognized that genetic factors substantially contribute to individual differences in HPA-axis reactivity,2, 3, 4 the search for robust associations with specific genetic variants has proven challenging. Among others,5,6 a large number of neuroendocrine studies7, 8, 9, 10, 11, 12, 13, 14 have investigated a 43-bp insertion/deletion polymorphism (5-HTTLPR) in the serotonin transporter gene (SLC6A4) which comprises a short (S) and a long (L) allelic variant.15 This line of research is motivated by a strong biological and clinical rationale. First, the S allele has been repeatedly linked to a lower transcriptional efficiency of the SLC6A4 gene15,16 and thus potentially alters serotonergic modulation of HPA-axis activity.17,18 Second, the debate about whether the S allele conveys vulnerability to depression upon exposure to environmental adversity19, 20, 21, 22 led to a growing interest in the biological pathways mediating genotype-dependent stress sensitivity. Consequently, several experimental studies have explored the association of 5-HTTLPR and cortisol stress reactivity to standardized laboratory stressors.7, 8, 9, 10, 11, 12, 13, 14 In support of the stress sensitivity hypothesis, a recent meta-analysis confirms an overall increased HPA-axis reactivity in individuals carrying two copies of the S allele.23 However, the size of this effect was small with substantial variation between individual study outcomes, which highlights the need to explore additional sources of variance contributing to observed inconsistencies.
A more powerful and comprehensive approach in neuroendocrine candidate gene studies might be to account for functional variation within SLC6A4 both on a genetic and epigenetic level. Previous research has identified cytosine methylation within a 799-bp promoter-associated CpG island of the SLC6A4 gene as a functionally relevant marker of reduced serotonin transporter (SERT) expression.24, 25, 26, 27 Given that SLC6A4 methylation profiles appear to be sensitive to early-life stress in several studies,26,28, 29, 30, 31 they reflect a putative pathway explaining how environmental adversity might translate into stress and disease susceptibility. Indeed, initial evidence suggests an association of SLC6A4 methylation levels measured in peripheral cells and depression,25,30,32,33 in one study dependent on 5-HTTLPR genotype.24 It is thus conceivable that in addition to genetic variation, epigenetic changes within SLC6A4 may also relate to differences in endocrine stress reactivity. To date, only one recent study on 28 monozygotic twin pairs discordant for bullying victimization has examined neuroendocrine correlates of epigenetic changes in SLC6A4.31 This study reports increased SLC6A4 methylation at 1 out of the 12 CpG sites studied in bullied twins which in turn was related to a blunted cortisol stress response. Such epigenetic recalibration of the neuroendocrine system presumably reflects a flexible adaption to environmental demands, but may also convey disease vulnerability if the programmed response is not adequate anymore.34
The current study is the first to investigate combined effects of functional genetic and epigenetic variation in the SLC6A4 gene on endocrine stress reactivity. Our major aim was to test whether SLC6A4 methylation modulates allele-specific cortisol response patterns and whether a larger portion of variance can be explained when both factors are taken into account. Respective findings are used to evaluate whether studying epigenetic marks might help to detect more robust effects in neuroendocrine and clinical studies investigating the 5-HTTLPR stress sensitivity hypothesis.
Materials and methods
Sample and procedure
The study sample comprised two hundred healthy participants (n=100 females) aged 18–30 years. One hundred and thirty three participants from a previous study investigating associations between environmental adversity, SLC6A4 methylation and mRNA expression patterns were re-invited for the present study.27 This initial sample was extended by another sixty seven individuals who were exposed to exactly the same procedure. We included only Caucasians who were native German speakers. Exclusion criteria were current or past mental and/or physical diseases, medication intake (for example, psychotropic drugs, substances known to influence HPA-axis activity), pregnancy, an irregular menstrual cycle and a body mass index (BMI) <17 or >30. After a structured telephone interview screening for exclusion criteria, participants were invited to the first session, which comprised the diagnostic interview for psychiatric disorders—short version (Mini-DIPS)35 to assess point and lifetime prevalence of axis I disorders on the basis of DSM IV criteria. Next, participants filled in a set of questionnaires, including a detailed checklist on chronic physical diseases and medication status, the Beck Depression Inventory (BDI-II36), the short form of the childhood trauma questionnaire (CTQ37,38) for the assessment of childhood maltreatment and the life stressor checklist—revised (LSC-R39,40) to obtain information on traumatic/stressful life events within the past 5 years. Finally, blood samples were drawn into EDTA tubes (Sarstedt, Nümbrecht, Germany) for DNA extraction and stored at −20 °C for no more than 6 months. The experimental sessions were scheduled on a second appointment carried out within close succession. The study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of the Technische Universität Dresden. Participants provided written informed consent and received a monetary reward for participation.
Standardized laboratory stress test: TSST
All participants were exposed to the Trier Social Stress Test (TSST),41 a standardized protocol known to reliably elicit robust cortisol increases.42 The cortisol response to the TSST is characterized by substantial intraindividual stability across repeated test session along with a moderate heritability,2,3 which highlights the use of this procedure for (epi)genetic association studies. In short, the TSST consists of a public speaking (5 min) and a mental arithmetic (5 min) task performed in front of two evaluating panelists. During the experimental procedure, seven saliva samples were collected before onset of the TSST (after a 30 min resting period) as well as 1, 10, 20, 30, 45 and 60 min after stress exposure. Experimental sessions started between 1330 and 1500 hours to reduce variability in the circadian cortisol rhythm. Participants were instructed to refrain from physical exercising, smoking, eating and drinking anything but water 1 h before test sessions. For females, TSST appointments were scheduled during the second half of the menstrual cycle only (corresponding to the luteal phase in women free of oral contraceptives). To avoid creating a highly selective sample within this age group, smokers and oral contraceptive users were not excluded but these variables were treated as potential confounders in statistical analyses.
5-HTTLPR genotyping
DNA was extracted from EDTA whole blood samples by means of standard commercial extraction kits (High Pure PCR Template Preparation Kit; Roche, Mannheim, Germany) in a MagNA Pure LC System (Roche). Genotyping was performed according to a previously published protocol.7 Participants were further genotyped for an A/G single-nucleotide polymorphism (rs25531) within the length polymorphism which results in the distinction between the variants LA and LG, with the latter being functionally similar to the S allele.16 This allows for conducting analyses based on the 5-HTTLPR/rs25531 mini-haplotype by comparing low [LG/S] and high [LA] expressing variants.
Bisulfite pyrosequencing
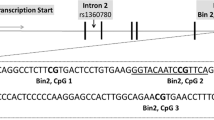
Quantitative methylation analysis of 83 CpG sites within a previously described43 799-bp CpG island in SLC6A4 (Supplementary Information 1) was performed by Varionostic GmbH (Ulm, Germany, http://www.varionostic.de). Genomic DNA extracted from EDTA whole blood was bisulfite-treated using the EZ DNA Methylation Gold Kit (Zymo Research, Range, CA, USA) and subsequent pyrosequencing was performed on the Q24/ID System. A detailed protocol has been published elsewhere.27 Mean SLC6A4 methylation levels across the CpG island were calculated for those participants where methylation values for at least 90% of the 83 CpG sites passed a strict quality control, which led to the exclusion of 14 individuals. Consequently, all subsequent analyses on SLC6A4 methylation refer to a sample of N=186.
Hormone assays
Saliva samples were collected by means of cotton swabs (Salivettes, Sarstedt). Participants were instructed to chew on the swabs for 3 min to stimulate saliva flow. Cotton rolls were then transferred to plastic containers and stored at −20 °C. For cortisol analyses, saliva samples were thawed and centrifuged at 3000 r.p.m. for 3 min. Salivary-free cortisol levels were measured using commercially available chemiluminescence-immunoassays (CLIA; IBL, Hamburg, Germany) with intra- and inter-assay precision of 3.0 and 4.2%.
Statistical analyses
Statistical analyses were conducted using SPSS (Version 21.0. IBM, Chicago, IL, USA). All statistical tests were two-tailed with alpha set at P<0.05. Initial group comparisons were conducted using χ2 tests and analyses of variance (ANOVA). Given that Kolmogorov–Smirnov testing indicated a deviation from a normal distribution for absolute cortisol values, all analyses were based on natural log-transformed values. For descriptive purposes, mean data in tables and figures are presented in original units. To test for effects of 5-HTTLPR, SLC6A4 methylation (mean methylation levels across the 83 CpG sites) and their interaction on cortisol reactivity, mixed-design analyses of covariance with repeated measures (cortisol samples) and baseline cortisol values as covariate were conducted. Regarding 5-HTTLPR as between-subject factor, analyses of covariance were calculated on a genotype level (SS vs SL vs LL) and for completeness also according to an L or S allele dominant model. Equivalent analyses were conducted on the basis of the 5-HTTLPR/rs25531 mini-haplotype by comparing the load of high (LA) and low (LG,S) expressing alleles (SS, SLG, LGLG vs SLA, LGLA vs LALA). Regarding mean SLC6A4 methylation as between-subject factor, participants were assigned to a ‘low’ and ‘high’ methylation group by median split to illustrate associations with cortisol response patterns over time in a repeated-measure design. Additional analyses were conducted using the area under the cortisol curve with respect to increase (AUCI)44 as the dependent variable to investigate Pearson correlations with SLC6A4 methylation and to calculate linear regression analyses testing for 5-HTTLPR × SLC6A4 methylation interaction. In accordance with previous research,45 we identified sex (males>females), smoking status (nonsmoker>smoker) and oral contraceptives (nonusers>users) as variables associated with cortisol reactivity (all P-values⩽0.007), whereas no associations with age, BMI, depression score, childhood maltreatment and recent stress/trauma exposure were seen (all P-values⩾0.134). Although being equally distributed across genotype/methylation groups (Table 1), additional analyses were calculated controlling for sex, smoking status and oral contraceptives to ensure robustness of the current results.
Results
Sample characteristics
Table 1 shows sample characteristics for participants grouped by 5-HTTLPR genotype or SLC6A4 methylation level. There was no significant deviation from Hardy–Weinberg equilibrium using bi-allelic (x2(1)=0.82, P=0.37) or the 5-HTTLPR/rs25531 mini-haplotype (x2(3)=2.33, P=0.13) classification. As previously reported in a subsample,27 we observed substantial interindividual variation regarding absolute SLC6A4 methylation levels (Supplementary Information 1). Mean SLC6A4 methylation levels were comparable between groups separated by 5-HTTLPR genotype (F1,182=0.12, P=0.89). Participants grouped by 5-HTTLPR genotype or SLC6A4 methylation level did not differ regarding sex, age, BMI, smoking status, oral contraceptive use depression score, childhood maltreatment and recent trauma/stress exposure (all P-values⩾0.11).
Main effects of 5-HTTLPR and SLC6A4 methylation on cortisol stress reactivity
The TSST induced significant increases in salivary cortisol across the overall sample (F6,1194=198.95, P⩽0.001, η2=0.50). Table 2 displays cortisol levels in response to the TSST as a function of 5-HTTLPR and SLC6A4 methylation levels. Despite a nominal trend for higher HPA-axis reactivity in S allele carriers, analyses of covariance revealed no significant effect of 5-HTTLPR on cortisol response patterns when comparing individuals on a genotype (genotype: F2,196=0.98, P=0.378; genotype × time: F12,1176=1.45, P=0.137) or allele-specific level (S allele dominant model, genotype: F1,197=1.95, P=0.165, genotype × time F6,1182=1.29, P=0.26; L allele dominant model, genotype: F1,197=0.90, P=0.771, genotype × time: F6,1182=1.32, P=0.244). When reanalyzing the data on the basis of the 5-HTTLPR/rs25531 mini-haplotype, we observed a trend for carriers of two low-expressing alleles to have higher cortisol responses compared with individual with at least one high-expressing allele (genotype: F1,197=1.752, P=0.187, genotype × time: F6,1182=2.05, P=0.056, η2=0.01). The latter effect reached significance when controlling for sex, smoking status and use of oral contraceptives (genotype × time: F6,1164=2.34, P=0.03, η2=0.01), however, explained variance in cortisol reactivity was small. Again, no differences in cortisol response patterns were found on a genotype level (genotype: F2,196=1.03, P=0.361; genotype × time: F12,1176=1.39, P=0.162) or when applying a dominant model for the high-expressing alleles (genotype: F1,197=0.82, P=0.366; genotype × time: F6,1182=1.08, P=0.373).
Regarding main effects of epigenetic modifications in SLC6A4, analyses of covariance revealed no significant differences in cortisol response patterns between individuals characterized by high or low mean SLC6A4 methylation levels (methylation: F1,183=0.41, P=0.839, methylation × time: F6,1098=0.36, P=0.905). Accordingly, mean SLC6A4 methylation levels were unrelated to the cortisol AUCI (r=−0.018, P=0.805). We further conducted exploratory analyses on the level of individual CpG sites, which generally confirmed the absence of a significant association of SLC6A4 methylation and cortisol stress reactivity. Methylation levels at 3 out of 83 CpG sites (CpG35: r=0.14, P=0.045, CpG51: r=−0.19, P=0.008, CpG64: r=−0.16, P=0.031) were nominally associated with cortisol AUCI (before Bonferroni adjustment). However, these associations are likely to result from chance (P(i⩾3|α=0.05, n=83)=0.79).
Interaction of genetic and epigenetic variation in the SLC6A4 gene on cortisol stress reactivity
The major study finding suggests a significant interaction of 5-HTTLPR and SLC6A4 methylation status on cortisol stress reactivity (genotype × methylation: F2,179=3.66, P=0.028, η2=0.04; genotype × methylation × time: F12,1074=2.59, P=0.002, η2=0.03; Figure 1). Post hoc analysis revealed a significant effect of 5-HTTLPR when SLC6A4 methylation was low, characterized by a dose-dependent effect of the S allele being associated with higher cortisol levels (genotype: F2,89=4.17, P=0.019, η2=0.09; genotype × time F12,534=3.41, P⩽0.001, η2=0.07, Figure 1a). No such effect could be observed in the high methylation group where a moderate cortisol response occurred across all genotype groups, indicating that high SLC6A4 methylation prevents genotype-specific effects (genotype: F2,89=0.72, P=0.492, genotype × time: F12,534=0.74, P=0.711, Figure 1b). We further conducted linear regression analyses using cortisol AUCi as the dependent variable which confirmed a significant 5-HTTLPR × SLC6A4 methylation interaction (t=2.94, β=0.95, P=0.004). Including this 5-HTTLPR × SLC6A4 methylation interaction term in the regression model (F3,182=3.10, P=0.028, R2=0.049) incrementally increased the portion of variance explained by an additive model (F2,183=0.32, P=0.730, R2=0.003).

5-HTTLPR (bi-allelic classification) × SLC6A4 methylation interaction on cortisol stress reactivity. (a) Mean (±s.e.m.) salivary cortisol levels in response to the Trier Social Stress Test as a function of 5-HTTLPR genotype (bi-allelic classification) in individuals displaying low levels of mean SLC6A4 methylation. (b) Mean (±s.e.m.) salivary cortisol levels in response to the Trier Social Stress Test as a function of 5-HTTLPR genotype (bi-allelic classification) in individuals displaying high levels of mean SLC6A4 methylation.
A comparable 5-HTTLPR × SLC6A4 methylation interaction occurred when analyses were conducted on the basis of the 5-HTTLPR/rs25531 mini-haplotype (genotype × methylation: F2,179=3.21, P=0.043, η2=0.04, genotype × methylation × time: F12,1074=1.89, P=0.032, η2=0.02, Figure 2). Again, a dose-dependent association between the low-expression alleles and cortisol stress reactivity was found when SLC6A4 methylation was low (genotype: F2,89=4.00, P=0.022, η2=0.08; genotype × time F12,534=2.86, P⩽0.001, η2=0.06, Figure 2a), whereas no genotype-dependent differences occurred in the high methylation group (genotype: F2,89=0.25, P=0.778, genotype × time: F12,534=0.43, P=0.953, Figure 2b). Accordingly, linear regression analysis revealed a significant 5-HTTLPR × SLC6A4 methylation interaction on cortisol AUCI (t=2.25, β=0.72, P=0.025). Again, inclusion of this interaction term in the regression model (F3,182=2.30, P=0.412, R2=0.037) incrementally increased the portion of variance in cortisol AUCI explained by an additive model (F2,183=0.89, P=0.412, R2=0.010).

5-HTTLPR/rs25531 × SLC6A4 methylation interaction on cortisol stress reactivity. (a) Mean (±s.e.m.) salivary cortisol levels in response to the Trier Social Stress Test as a function of 5-HTTLPR/rs25531 genotype in individuals displaying low levels of mean SLC6A4 methylation. (b) Mean (±s.e.m.) salivary cortisol levels in response to the Trier Social Stress Test as a function of 5-HTTLPR/rs25531 genotype in individuals displaying high levels of mean SLC6A4 methylation.
Comparable results regarding a significant interaction of 5-HTTLPR and SLC6A4 methylation were achieved when reanalyzing the data by including sex, smoking status or oral contraceptives intake as covariates in the statistical model.
Discussion
This is the first study to report a significant interaction of functional genetic and epigenetic variation at the SLC6A4 locus on endocrine stress reactivity in a sample of healthy, young adults. Specifically, methylation patterns within a promoter-associated CpG island of the SLC6A4 gene were found to moderate the association of 5-HTTLPR and cortisol reactivity to psychosocial stress. For individuals displaying low levels of SLC6A4 methylation, the S allele relates to increased cortisol stress reactivity in a dose-dependent fashion, whereas no such effect occurred when SLC6A4 methylation was high. As expected from meta-analytic results,23 the effect of 5-HTTLPR on cortisol stress reactivity appeared to be small and mostly falls below significance in the overall sample, while explaining 7–9% of the variance in case of low SLC6A4 methylation. Accounting for epigenetic modifications in SLC6A4 might thus allow for a more robust detection of effects when evaluating the 5-HTTLPR stress sensitivity hypothesis in neuroendocrine and clinical studies.
Given that epigenetic modifications are discussed as a key process mediating long-term changes of HPA-axis regulation in response to environmental exposure,34 the present findings may reflect a molecular substrate of G × E interaction. In line with G × E research on depression,19, 20, 21 accumulating evidence suggests that heightened neural and endocrine stress sensitivity associated with the 5-HTTLPR S allele is most pronounced upon environmental adversity.7,46,47 More generally, these findings concur with twin studies indicating that heritability of cortisol levels at rest48 and in response to stress4 substantially varies as a function of early adversity. Although genetic factors were found to account for a considerable amount of variance in HPA-axis reactivity when familiar adversity was low, no heritable component could be detected in a high stress environment.4 Our finding of a stress-relevant genetic variant being less important upon epigenetic modifications could be discussed as a specific example and mechanism here, given that individual methylation profiles partly result from differential environmental exposure.34,49 One intriguing advantage of investigating functional epigenetic markers in SLC6A4 could be that they presumably reflect the net effects of a broad range of those environmental influences relevant for long-term changes in outcome measures under serotonergic control. This is of particular importance as the inability to capture the relevant type and timing of adversity (with the appropriate method) has been identified as a crucial source of inconsistencies in previous G × E research.50,51 A series of studies have recently started to link several types of early adversity with increased SLC6A4 methylation,26,28, 29, 30, 31 however, no specific CpG site has yet been consistently identified across individual studies. As reported elsewhere, we could not replicate such associations for prenatal stress and childhood maltreatment,27 indicating that these specific stressors are unlikely to account for the moderating role of SLC6A4 methylation in our study. Although the precise environmental (and genetic) correlates of SLC6A4 methylation are largely unknown, future studies should elucidate whether the use of epigenetic markers instead of specific life events will yield more consistent results in G × E research.
The finding of SLC6A4 methylation compensating for increased cortisol reactivity in S allele carriers also raises the possibility that epigenetic modifications reflect an adaptive fine-tuning of the genetically influenced stress response. In favor of this assumption, our study reveals that increased SLC6A4 methylation was associated with a relatively uniform expression of a normal, moderately sized cortisol stress response across different 5-HTTLPR genotype groups. Such moderate cortisol reactivity is often considered as an adaptive response preparing the organism to successfully cope with environmental challenges,52 while both hyper- and hypoactivity of the HPA-axis are frequent correlates of psychopathology.53,54 Indeed, this hypothesis closely concurs with a recent report suggesting that vulnerability of S/S carriers for developing psychological problems in response to stress may be reduced by increased SLC6A4 methylation.55 In this study, the S/S genotype was associated with more unresolved responses to traumatization, but only in the case of lower methylation within the promoter-associated SLC6A4 CpG island.55 Seemingly conflicting evidence for SLC6A4 methylation being an adaptive process is provided by first studies linking epigenetic modifications in SLC6A4 to vulnerability for stress-related psychopathology.56 Most of these studies found higher mean or site-specific SLC6A4 methylation in depressed patients compared with healthy controls,25,30,33 however, opposite results have also been obtained.32 Still, it remains difficult to draw firm conclusions about whether increased SLC6A4 methylation either reflects the body’s attempt to adapt to environmental adversity or constitutes a risk factor for psychopathology. As (specific) trauma exposure per se may relate to higher SLC6A4 methylation26,28, 29, 30, 31 and is also more frequently observed in depressed patients,22,57 including both traumatized and non-traumatized healthy controls might help to resolve this question.
Future studies should provide further insights into the underlying mechanisms by which genetic and epigenetic modification in the SLC6A4 gene interact to shape stress-relevant pathways. From a molecular perspective, one might have expected that SLC6A4 methylation further promotes increased stress sensitivity in S allele carriers, given that both the S allele15,16,27 and SLC6A4 methylation24, 25, 26, 27 relate to lower serotonin transporter expression. Although our findings are not supportive of such additive effects with regard to cortisol reactivity, it is important to note that a disruption of serotonin transporter functioning may have very distinct impact on the organism during different developmental stages.58 One example refers to the ‘SSRI paradox’, where early pre- and postnatal blockage of the serotonin transporter by SSRI (selective serotonin reuptake inhibitors) was found to induce sustained depressogenic effects in rodents.59, 60, 61 This led to the assumption that 5-HTTLPR primarily exerts its unfavorable effects during early neurodevelopment when deficient serotonin transporter functioning in S allele carriers might disrupt maturation of stress-relevant brain circuits.58 In contrast, individual differences in SLC6A4 methylation might gradually evolve as a function of environmental exposure, thereby possibly exerting maximum impact during a later time period. Unlike the unfavorable effects on stress sensitivity observed during early brain maturation, inhibition of serotonin transporter functioning (for example, via SSRI) is known to effectively reduce symptoms of depression and HPA-axis hyperactivity later in life.62,63 Despite comparable effects on gene expression, it is thus conceivable that genetic and epigenetic changes in SLC6A4 operate during different time windows to exert divergent effects on the stress response system.
Several limitations should be acknowledged for the present study. First, the current results rely on peripheral measures of SLC6A4 methylation, which may not generalize to the central nervous system. Nonetheless, the usefulness of peripheral epigenetic biomarkers has been increasingly recognized in psychiatric research56 given that methylation profiles,64 as well as interindividual variation in those epigenetic signatures,65 are significantly correlated across peripheral and neural cells. Furthermore, accumulating evidence suggests that environmentally induced epigenetic changes appear to be system-wide.66,67 For SLC6A4 in particular, a recent imaging study indicates peripheral SLC6A4 methylation to be an informative marker of human brain serotonin synthesis.68 Second, we chose to analyze whole blood as this procedure does not require transformations known to modify DNA methylation,69 however, the heterogeneous mixture of cell types may constitute a potential confound here. Finally, our finding of SLC6A4 methylation profiles moderating the link between 5-HTTLPR and cortisol stress reactivity remains preliminary until independent replication is available.
References
Chrousos GP . Stress and disorders of the stress system. Nat Rev Endocrinol 2009; 5: 374–381.
Federenko IS, Nagamine M, Hellhammer DH, Wadhwa PD, Wust S . The heritability of hypothalamus pituitary adrenal axis responses to psychosocial stress is context dependent. J Clin Endocrinol Metab 2004; 89: 6244–6250.
Kirschbaum C, Wust S, Faig HG, Hellhammer DH . Heritability of cortisol responses to human corticotropin-releasing hormone, ergometry, and psychological stress in humans. J Clin Endocrinol Metab 1992; 75: 1526–1530.
Ouellet-Morin I, Boivin M, Dionne G, Lupien SJ, Arseneault L, Barr RG et al. Variations in heritability of cortisol reactivity to stress as a function of early familial adversity among 19-month-old twins. Arch Gen Psychiatry 2008; 65: 211–218.
Derijk RH . Single nucleotide polymorphisms related to HPA axis reactivity. Neuroimmunomodulation 2009; 16: 340–352.
Kumsta R, Entringer S, Koper JW, van Rossum EF, Hellhammer DH, Wust S . Sex specific associations between common glucocorticoid receptor gene variants and hypothalamus-pituitary-adrenal axis responses to psychosocial stress. Biol Psychiatry 2007; 62: 863–869.
Alexander N, Kuepper Y, Schmitz A, Osinsky R, Kozyra E, Hennig J . Gene-environment interactions predict cortisol responses after acute stress: implications for the etiology of depression. Psychoneuroendocrinology 2009; 34: 1294–1303.
Bouma E, Riese H, Nederhof E, Ormel J, Oldehinkel A . No replication of genotype effect of 5-HTTLPR on cortisol response to social stress in larger adolescent sample. Biol Psychiatry 2010; 68: e33–e34.
Dougherty LR, Klein DN, Congdon E, Canli T, Hayden EP . Interaction between 5-HTTLPR and BDNF Val66Met polymorphisms on HPA axis reactivity in preschoolers. Biol Psychol 2010; 83: 93–100.
Gotlib IH, Joormann J, Minor KL, Hallmayer J . HPA axis reactivity: a mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biol Psychiatry 2008; 63: 847–851.
Mueller A, Armbruster D, Moser DA, Canli T, Lesch KP, Brocke B et al. Interaction of serotonin transporter gene-linked polymorphic region and stressful life events predicts cortisol stress response. Neuropsychopharmacology 2011; 36: 1332–1339.
Verschoor E, Markus CR . Effects of acute psychosocial stress exposure on endocrine and affective reactivity in college students differing in the 5-HTTLPR genotype and trait neuroticism. Stress 2011; 14: 407–419.
Way BM, Taylor SE . The serotonin transporter promoter polymorphism is associated with cortisol response to psychosocial stress. Biol Psychiatry 2010; 67: 487–492.
Wust S, Kumsta R, Treutlein J, Frank J, Entringer S, Schulze TG et al. Sex-specific association between the 5-HTT gene-linked polymorphic region and basal cortisol secretion. Psychoneuroendocrinology 2009; 34: 972–982.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996; 274: 1527–1531.
Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD et al. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet 2006; 78: 815–826.
Fuller RW . Serotonin receptors and neuroendocrine responses. Neuropsychopharmacology 1990; 3: 495–502.
Porter RJ, Gallagher P, Watson S, Young AH . Corticosteroid-serotonin interactions in depression: a review of the human evidence. Psychopharmacology 2004; 173: 1–17.
Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE . Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry 2010; 167: 509–527.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301: 386–389.
Karg K, Burmeister M, Shedden K, Sen S . The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry 2011; 68: 444–454.
Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA 2009; 301: 2462–2471.
Miller R, Wankerl M, Stalder T, Kirschbaum C, Alexander N . The serotonin transporter gene-linked polymorphic region (5-HTTLPR) and cortisol stress reactivity: a meta-analysis. Mol Psychiatry 2013; 18: 1018–1024.
Olsson CA, Foley DL, Parkinson-Bates M, Byrnes G, McKenzie M, Patton GC et al. Prospects for epigenetic research within cohort studies of psychological disorder: a pilot investigation of a peripheral cell marker of epigenetic risk for depression. Biol Psychol 2010; 83: 159–165.
Philibert RA, Sandhu H, Hollenbeck N, Gunter T, Adams W, Madan A . The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am J Med Genet B 2008; 147b: 543–549.
Vijayendran M, Beach SR, Plume JM, Brody GH, Philibert RA . Effects of genotype and child abuse on DNA methylation and gene expression at the serotonin transporter. Front Psychiatry 2012; 3: 55.
Wankerl M, Miller R, Kirschbaum C, Hennig J, Stalder T, Alexander N . Effects of genetic and early environmental risk factors for depression on serotonin transporter expression and methylation profiles. Transl Psychiatry 2014; 4: e402.
Beach SR, Brody GH, Todorov AA, Gunter TD, Philibert RA . Methylation at SLC6A4 is linked to family history of child abuse: an examination of the Iowa Adoptee sample. Am J Med Genet B 2010; 153b: 710–713.
Beach SR, Brody GH, Todorov AA, Gunter TD, Philibert RA . Methylation at 5HTT mediates the impact of child sex abuse on women's antisocial behavior: an examination of the Iowa adoptee sample. Psychosom Med 2011; 73: 83–87.
Kang HJ, Kim JM, Stewart R, Kim SY, Bae KY, Kim SW et al. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog Neuropsychopharmacol Biol Psychiatry 2013; 44: 23–28.
Ouellet-Morin I, Wong CC, Danese A, Pariante CM, Papadopoulos AS, Mill J et al. Increased serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response to stress in childhood: a longitudinal study of discordant monozygotic twins. Psychol Med 2013; 43: 1813–1823.
Devlin AM, Brain U, Austin J, Oberlander TF . Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS One 2010; 5: e12201.
Zhao J, Goldberg J, Bremner JD, Vaccarino V . Association between promoter methylation of serotonin transporter gene and depressive symptoms: a monozygotic twin study. Psychosom Med 2013; 75: 523–529.
Szyf M . DNA methylation, behavior and early life adversity. J Genet Genomics 2013; 40: 331–338.
Margraf J . Entstehung und Handhabung des Mini-DIPS. Springer: Berlin Heidelberg, Germany, 1994.
Hautzinger M Keller F Kühner C . Beck Depressions-Inventar (BDI-II). Revision. Harcourt Test Services: Frankfurt/Main, Germany, 2006.
Bernstein DP, Stein JA, Newcomb MD, Walker E, Pogge D, Ahluvalia T et al. Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl 2003; 27: 169–190.
Wingenfeld K, Spitzer C, Mensebach C, Grabe HJ, Hill A, Gast U et al. [The German Version of the Childhood Trauma Questionnaire (CTQ):Preliminary Psychometric Properties.]. Psychother Psychosom Med Psychol 2010; 60: e13.
Wolfe J Kimerling R . Gender issues in the assessment of posttraumatic stress disorder. In: Wilson JR, Keane TM (eds). Assessing Psychological Trauma and PTSD. Guilford: New York, NY, USA, 1997. 192–238.
Ungerer O, Deter HC, Fikentscher E, Konzag TA . [Improved diagnostics of trauma-related disease through the application of the Life-Stressor Checklist]. Psychother Psychosom Med Psychol 2010; 60: 434–441.
Kirschbaum C, Pirke KM, Hellhammer DH . The 'Trier Social Stress Test'—a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology 1993; 28: 76–81.
Dickerson SS, Kemeny ME . Acute stressors and cortisol responses: a theoretical integration and synthesis of laboratory research. Psychol Bull 2004; 130: 355–391.
Philibert R, Madan A, Andersen A, Cadoret R, Packer H, Sandhu H . Serotonin transporter mRNA levels are associated with the methylation of an upstream CpG island. Am J Med Genet B Neuropsychiatr Genet 2007; 144b: 101–105.
Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH . Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003; 28: 916–931.
Kudielka BM, Hellhammer DH, Wust S . Why do we respond so differently? Reviewing determinants of human salivary cortisol responses to challenge. Psychoneuroendocrinology 2009; 34: 2–18.
Alexander N, Klucken T, Koppe G, Osinsky R, Walter B, Vaitl D et al. Interaction of the serotonin transporter-linked polymorphic region and environmental adversity: increased amygdala-hypothalamus connectivity as a potential mechanism linking neural and endocrine hyperreactivity. Biol Psychiatry 2012; 72: 49–56.
Canli T, Qiu M, Omura K, Congdon E, Haas BW, Amin Z et al. Neural correlates of epigenesis. Proc Natl Acad Sci USA 2006; 103: 16033–16038.
Ouellet-Morin I, Dionne G, Perusse D, Lupien SJ, Arseneault L, Barr RG et al. Daytime cortisol secretion in 6-month-old twins: genetic and environmental contributions as a function of early familial adversity. Biol Psychiatry 2009; 65: 409–416.
Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K et al. Prospects for epigenetic epidemiology. Am J Epidemiol 2009; 169: 389–400.
Uher R, McGuffin P . The moderation by the serotonin transporter gene of environmental adversity in the aetiology of mental illness: review and methodological analysis. Mol Psychiatry 2008; 13: 131–146.
Uher R, McGuffin P . The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update. Mol Psychiatry 2010; 15: 18–22.
McEwen BS . Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 2007; 87: 873–904.
Lopez-Duran NL, Kovacs M, George CJ . Hypothalamic-pituitary-adrenal axis dysregulation in depressed children and adolescents: a meta-analysis. Psychoneuroendocrinology 2009; 34: 1272–1283.
Morris MC, Compas BE, Garber J . Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: a systematic review and meta-analysis. Clin Psychol Rev 2012; 32: 301–315.
van IJzendoorn MH, Caspers K, Bakermans-Kranenburg MJ, Beach SR, Philibert R . Methylation matters: interaction between methylation density and serotonin transporter genotype predicts unresolved loss or trauma. Biol Psychiatry 2010; 68: 405–407.
Klengel T, Pape J, Binder EB, Mehta D . The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology 2014; 80c: 115–132.
Chapman DP, Whitfield CL, Felitti VJ, Dube SR, Edwards VJ, Anda RF . Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord 2004; 82: 217–225.
Homberg JR, Schubert D, Gaspar P . New perspectives on the neurodevelopmental effects of SSRIs. Trends Pharmacol Sci 2010; 31: 60–65.
Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA . Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science 2004; 306: 879–881.
Lisboa SF, Oliveira PE, Costa LC, Venancio EJ, Moreira EG . Behavioral evaluation of male and female mice pups exposed to fluoxetine during pregnancy and lactation. Pharmacology 2007; 80: 49–56.
Popa D, Lena C, Alexandre C, Adrien J . Lasting syndrome of depression produced by reduction in serotonin uptake during postnatal development: evidence from sleep, stress, and behavior. J Neurosci 2008; 28: 3546–3554.
Nickel T, Sonntag A, Schill J, Zobel AW, Ackl N, Brunnauer A et al. Clinical and neurobiological effects of tianeptine and paroxetine in major depression. J Clin Psychopharmacol 2003; 23: 155–168.
Hinkelmann K, Moritz S, Botzenhardt J, Muhtz C, Wiedemann K, Kellner M et al. Changes in cortisol secretion during antidepressive treatment and cognitive improvement in patients with major depression: a longitudinal study. Psychoneuroendocrinology 2012; 37: 685–692.
Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18: 4808–4817.
Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13: R43.
Lee RS, Tamashiro KL, Yang X, Purcell RH, Harvey A, Willour VL et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 2010; 151: 4332–4343.
Provencal N, Suderman MJ, Guillemin C, Massart R, Ruggiero A, Wang D et al. The signature of maternal rearing in the methylome in rhesus macaque prefrontal cortex and T cells. J Neurosci 2012; 32: 15626–15642.
Wang D, Szyf M, Benkelfat C, Provencal N, Turecki G, Caramaschi D et al. Peripheral SLC6A4 DNA methylation is associated with in vivo measures of human brain serotonin synthesis and childhood physical aggression. PLoS One 2012; 7: e39501.
Aberg K, van den Oord EJ . Epstein-barr virus transformed DNA as a source of false positive findings in methylation studies of psychiatric conditions. Biol Psychiatry 2011; 70: e25–e26; author reply e27–28.
Acknowledgements
The current study was funded by a grant from the German Research Foundation to NA (AL 1484/2-1). We thank Maximilian Trompetter and Karolin Gruner for assisting in participant recruitment and blood draw. We are further grateful for the valuable laboratory work of Sarah Brand, Gabriele Arnold and Cornelia Meineke.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Alexander, N., Wankerl, M., Hennig, J. et al. DNA methylation profiles within the serotonin transporter gene moderate the association of 5-HTTLPR and cortisol stress reactivity. Transl Psychiatry 4, e443 (2014). https://doi.org/10.1038/tp.2014.88
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.88
This article is cited by
-
Prenatal exposure to environmental air pollution and psychosocial stress jointly contribute to the epigenetic regulation of the serotonin transporter gene in newborns
Molecular Psychiatry (2023)
-
No long-term effects of antenatal synthetic glucocorticoid exposure on epigenetic regulation of stress-related genes
Translational Psychiatry (2022)
-
Factors related to age at depression onset: the role of SLC6A4 methylation, sex, exposure to stressful life events and personality in a sample of inpatients suffering from major depression
BMC Psychiatry (2021)
-
The association of the 5-HTTLPR polymorphism and the response to different stressors in healthy males
Journal of Neural Transmission (2021)
-
Risk factors for suicide attempt: A population-based -genetic study from Telangana, India
Current Psychology (2021)
