
Abstract
Autism spectrum disorders (ASDs) now affect 1–2% of the children born in the United States. Hundreds of genetic, metabolic and environmental factors are known to increase the risk of ASD. Similar factors are known to influence the risk of schizophrenia and bipolar disorder; however, a unifying mechanistic explanation has remained elusive. Here we used the maternal immune activation (MIA) mouse model of neurodevelopmental and neuropsychiatric disorders to study the effects of a single dose of the antipurinergic drug suramin on the behavior and metabolism of adult animals. We found that disturbances in social behavior, novelty preference and metabolism are not permanent but are treatable with antipurinergic therapy (APT) in this model of ASD and schizophrenia. A single dose of suramin (20 mg kg−1 intraperitoneally (i.p.)) given to 6-month-old adults restored normal social behavior, novelty preference and metabolism. Comprehensive metabolomic analysis identified purine metabolism as the key regulatory pathway. Correction of purine metabolism normalized 17 of 18 metabolic pathways that were disturbed in the MIA model. Two days after treatment, the suramin concentration in the plasma and brainstem was 7.64 μM pmol μl−1 (±0.50) and 5.15 pmol mg−1 (±0.49), respectively. These data show good uptake of suramin into the central nervous system at the level of the brainstem. Most of the improvements associated with APT were lost after 5 weeks of drug washout, consistent with the 1-week plasma half-life of suramin in mice. Our results show that purine metabolism is a master regulator of behavior and metabolism in the MIA model, and that single-dose APT with suramin acutely reverses these abnormalities, even in adults.
Similar content being viewed by others

Introduction
Genetic,1, 2, 3 environmental4,5 and metabolic6 factors can contribute to the risk of autism to different extents in each affected child. Despite this etiologic heterogeneity, and the well-known clinical variations that make each child unique, clinical studies suggest that a common denominator may underlie the shared behavioral and cognitive features that define autism spectrum disorders (ASDs) as a group. For example, in a prospective study conducted by the Zimmerman group at the Kennedy Krieger Institute in 2007, 83% of children with autism spectrum disorders were found to improve transiently in association with fever.7 Improvements were lost with the resolution of the fever. This study showed that, despite the many different causes of ASD, the symptoms were not permanent and could be improved in a substantial fraction of children.
Transient improvements with fever have also been found in patients with certain forms of post-infection brain syndromes, movement disorders, dementia and schizophrenia in the early 1900s, although those early studies were made complicated by the use of live malarial parasites to produce the fevers.8,9 In contrast to these beneficial effects, when the exposure to serious infection happens before the onset of disease—during early development, and particularly during pregnancy—the metabolic changes associated with significant fever or infection are known to increase the risk of neurodevelopmental disorders in the offspring. These disorders include schizophrenia,10 ASDs,11 attention deficit/hyperactivity disorder,12 bipolar disorder,13 epilepsy14 and cerebral palsy.15 The nature and developmental timing of the exposure are important. Metabolism and mitochondrial function change adaptively during and after infection, and are well-known regulators of neurotransmission16 and synaptic plasticity.17 Collectively, these studies suggest that, despite many different causes, the symptoms of several neurodevelopmental disorders such as ASD, schizophrenia and bipolar disorder may have a metabolic basis and be acutely responsive to treatment using the right metabolic intervention.
The maternal immune activation (MIA) mouse model of neurodevelopmental disorders produces symptoms that are biologically similar to those of ASD18 and schizophrenia.19 Pregnant females that are exposed to a simulated viral infection by injection of the double-stranded RNA poly(Inosine:Cytosine) produce offspring with features of ASD20 and schizophrenia.21 Exposure to poly(IC) activates an evolutionarily conserved metabolic response to a threat called the cell danger response (CDR).22 Pathological persistence of the CDR, beyond the physical presence of the threat, has been observed in a variety of chronic disorders including ASDs.22 Purinergic signaling has been hypothesized to be a key regulator of the CDR;22 however, this has not yet been proven. In support of this hypothesis, we recently showed that antipurinergic therapy (APT) in the MIA mouse model corrected all of the behavioral, molecular and neuropathological abnormalities when weekly treatment with the antipurinergic drug suramin was begun at 1.5 months of age, near the age of reproductive maturity for mice.23 Significant reductions in mitochondrial oxygen consumption and body temperature were also found. However, comprehensive metabolomic analysis was not reported in that study.23
In the present study, we tested the hypothesis that the behavioral manifestations of the MIA model are a consequence of pathological persistence of the evolutionarily conserved CDR,22 and that the CDR is maintained by dysregulated purine metabolism and secondary abnormalities in purinergic signaling. We found that a single dose of the antipurinergic drug suramin given to adult animals about 6 months of age (21–27 weeks) produced the concerted correction of over 90% of the metabolic pathway disturbances, and all of the behavioral abnormalities that we tested in the MIA model. Six-month-old mice are the human biological age equivalents of about 30 years24 (see Materials and methods). After washout of the drug, these improvements were lost and the former abnormalities returned. These data show that purine metabolism and purinergic signaling represent a novel neurochemical switch that regulates both behavior and metabolism in the MIA model of neurodevelopmental disorders such as ASD and schizophrenia.
Materials and methods
Animals and husbandry
All studies were conducted at the University of California, San Diego (UCSD) in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) under the UCSD Institutional Animal Care and Use Committee-approved animal subjects protocols, and followed the National Institutes of Health Guidelines for the use of animals in research. Six- to eight-week-old C57BL/6J (strain no. 000664) mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA), given food and water ad libitum, identified by ear tags, and used to produce the timed matings. Animals were housed in a temperature- (22–24 °C) and humidity (40–55%)-controlled vivarium with a 12-h light–dark cycle (lights on at 0700 hours). Nulliparous dams were mated at 9–10 weeks of age. The sires were also 9–10 weeks of age. The human biological age equivalent for the C57BL/6J strain of laboratory mouse (Mus musculus) can be estimated from the following equation: 12 years for the first month, 6 years for the second month, 3 years for months 3–6 and 2.5 years for each month thereafter.24 Therefore, a 6-month-old mouse would be the biological equivalent of 30 years old (=12+6+3 × 4) on a human timeline.
Poly(IC) preparation and gestational exposure
To initiate the MIA model, pregnant dams were given two intraperitoneal injections of Poly(I:C) (Potassium salt; Sigma-Aldrich, St. Louis, MO, USA, Cat no. P9582; >99% pure; <1% mononucleotide content). These were quantified by UV spectrophotometry. One unit (U) of poly(IC) was defined as 1 absorbance unit at 260 nm. Typically, 1U=12 μg of RNA. 0.25 U/g [3 mg kg−1] of poly(IC) was given on E12.5 and 0.125 U g−1 (1.5 mg kg−1) on E17.5 as previously described.23 Contemporaneous control pregnancies were produced by timed matings and randomized assignment of pregnant dams to saline injection (5 μl g−1 intraperitoneally (i.p.)) on E12.5 and E17.5.
Postnatal handling and antipurinergic therapy (APT)
Offspring of timed matings were weaned at 3–4 weeks of age into cages of two to four animals. No mice were housed in isolation. Only males were evaluated in these studies. Littermates were identified by ear tags and distributed into different cages in order to minimize litter and dam effects. To avoid chance differences in groups selected for single-dose treatment, the saline and poly(IC) exposure groups were each balanced according to their social approach scores at 2.25 months. At 5.25 or 6.5 months of age, half the animals received a single injection of either saline (5 μl g−1 i.p.) or suramin (hexasodium salt, 20 mg kg−1 i.p.; Tocris Bioscience, Bristol, UK, Cat no. 1472). Beginning 2 days later, behaviors were evaluated as described below. After completing the behavioral measurements, half of the subjects were killed after a 5-week-washout period for measurement of suramin tissue levels. For acute suramin levels, the other half was injected at 7.75 months of age and killed 2 days later for tissue level determinations.
Behavioral testing
Behavioral testing began at 2.25 months (9 weeks) of age. Mice were tested in social approach, rotarod, t-maze test of spontaneous alternation and light–dark box test. If abnormalities were found, treatment with suramin or saline was given at 5.25 months (21 weeks) or 6.5–6.75 months (26–27 weeks) and the testing was repeated. Only male animals were tested.
Social approach
Social behavior was tested as social preference as previously described23 with minor modifications (see Full Methods in Supplementary Information; N=19–25, 2.25-month-old males per group before adult treatment with suramin. N=8–13, 6.5-month-old males per group).
T-Maze
Novelty preference was tested as spontaneous alternation behavior in the T-maze by a modification of the methods of Frye and Walf25 (see Full Methods Supplementary Information). N=19–25, 4-month-old males per group before adult treatment with suramin. N=8–13, 5.25-month-old males per group.
Rotarod
Sensorimotor coordination was tested as latency to fall on the rotarod as previously described23 (see Full Methods Supplementary Information; N=19–25, 2.5-month-old males per group before adult treatment with suramin. N=8–13, 6.75-month-old males per group).
Light–dark box
Certain anxiety-related and light-avoidance behaviors were tested in the light–dark box paradigm as previously described26 (see Full Methods Supplementary Information; N=19–25, 3.5-month-old males per group).
Absence of abnormal behaviors produced by suramin
This was assessed in the non-MIA control animals (indicated as the ‘Saline’ group in the pretreatment figures) that were injected with suramin as adults (indicated as the ‘Sal-Sur’ groups in the single-dose treatment figures) using each of the above behavioral paradigms.
Suramin quantitation
Tissue samples (brainstem, cerebrum and cerebellum) were ground into powder under liquid nitrogen in a pre-cooled mortar. Powdered tissue (15–50 mg) was weighed and mixed with the internal standard trypan blue to a final concentration of 5 μM (pmol mg−1) and incubated at room temperature for 10 min to permit metabolite interaction with binding proteins. Nine volumes of methanol:acetonitrile:H2O (43:43:16) pre-chilled to −20 °C was added to produce a final solvent ratio of 40:40:20, and the samples were deproteinated and macromolecules removed by precipitation on crushed ice for 30 min. The mixture was centrifuged at 16 000 g for 10 min at 4 °C and the supernatant was transferred to a new tube and kept at −80 °C for further LC-MS/MS (liquid chromatography-tandem mass spectrometry) analysis. For plasma, 90 μl was used, to which 10 μl of 50 μM stock of trypan blue was added to achieve an internal standard concentration of 5 μM. This was incubated at room temperature for 10 min to permit metabolite interaction with binding proteins, then extracted with 4 volumes (400 μl) of pre-chilled methanol:acetonitrile (50:50) to produce a final concentration of 40:40:20 (methanol:acetonitrile:H2O) and precipitated on ice for 10 min. Other steps were the same as for solid tissue extraction.
Suramin was analyzed on an AB SCIEX QTRAP 5500 triple quadrupole mass spectrometer equipped with a Turbo V electrospray ionization source, Shimadzu LC-20A UHPLC system, and a PAL CTC autosampler (AB SCIEX, Framingham, MA, USA). Ten microliters of extract were injected onto a Kinetix pentafluorophenyl column (150 × 2.1 mm, 2.6 μm; Phenomenex, Torrance, CA, USA) held at 30 °C for chromatographic separation. The mobile phase A was water with 20 mM ammonium acetate (NH4OAC; pH 7) and mobile phase B was methanol with 20 mM NH4OAC (pH 7). Elution was performed using the following gradient: 0 min—0% B, 15 min—100% B, 18 min—100% B, 18.1 min—0% B, 23 min—end. The flow rate was 300 μl min−1. All the samples were kept at 4 °C during analysis. Suramin and trypan blue were detected using scheduled multiple reaction monitoring (MRM) with a dwell time of 30 ms in negative mode and retention time window of 7.5–8.5 min for suramin and 8.4–9.4 min for trypan blue. MRM transitions for the doubly charged form of suramin were 647.0 m z−1 (Q1) precursor and 382.0 m z−1 (Q3) product. MRM transitions for trypan blue were 435.2 (Q1) and 185.0 (Q3). Absolute concentrations of suramin were determined for each tissue using a tissue-specific standard curve to account for matrix effects, and the peak area ratio of suramin to the internal standard trypan blue. The declustering potential, collision energy, entrance potential and collision exit potential were −104, −9.5,−32 and −16.9, and −144.58, −7, −57.8 and −20.94 for suramin and trypan blue, respectively. The electrospray ionization source parameters were set as follows: source temperature 500 °C; curtain gas 30; ion source gas 1, 35; ion source gas 2 35; spray voltage −4500 V. Analyst 1.6.1 was used for data acquisition and analysis. N=4–6 per tissue. Results are reported as means±s.e.m. in absolute μM (pmol μl−1) concentration for plasma, and pmol mg−1 wet weight for tissues.
Metabolomics
Broad-spectrum analysis of 478 targeted metabolites from 44 biochemical pathways in the plasma was performed by a modification of the methods described by Bajad and Shulaev.27 Only male animals that had been behaviorally evaluated were tested. Samples were analyzed on an AB SCIEX QTRAP 5500 triple quadrupole mass spectrometer equipped with a Turbo V electrospray ionization source, Shimadzu LC-20A UHPLC system and a PAL CTC autosampler (AB SCIEX). Whole blood was collected 2 daysafter a single dose of suramin (20 mg kg−1 i.p.) or saline (5 μl g−1 i.p.) from animals that were lightly anesthetized with isoflurane (Med-Vet International, Mettawa, IL, USA, Cat no. RXISO-250) in a drop jar into BD Microtainer tubes containing lithium heparin (Becton Dickinson, San Diego, CA, USA, Ref no. 365971) by submandibular vein lancet.28 Plasma was separated by centrifugation at 600 g × 5 min at 20 °C within 1 h of collection. Fresh lithium–heparin plasma was transferred to labeled tubes for storage at −80 °C for analysis. Typically, 45 μl of plasma was thawed on ice and transferred to a 1.7-ml Eppendorf tube. Two and one-half (2.5) microliters of a cocktail containing 35 commercial stable isotope internal standards (Supplementary Table S3) and 2.5 μl of 310 stable isotope internal standards that were custom-synthesized in Escherichia coli and Saccharomyces cerevisiae by metabolic labeling with 13C-glucose and 13C-bicarbonate were added, mixed and incubated for 10 min at 20 °C to permit small molecules and vitamins in the internal standards to associate with plasma-binding proteins. Macromolecules (protein, DNA, RNA and so on) were precipitated by extraction with 4 volumes (200 μl) of cold (−20 °C), acetonitrile:methanol (50:50) (LCMS grade, Cat no. LC015-2.5 and GC230-4, Burdick & Jackson, Honeywell, Muskegon, MI, USA), vortexed vigorously and incubated on crushed ice for 10 min, and then removed with centrifugation at 16 000 g × 10 min at 4 °C. The supernatants containing the extracted metabolites and internal standards in the resulting 40:40:20 solvent mix of acetonitrile:methanol:water were transferred to labeled cryotubes and stored at −80 °C for LC-MS/MS (liquid chromatography-tandem mass spectrometry) analysis.
LC-MS/MS analysis was performed by MRM under the Analyst v1.6.1 software control in both negative and positive modes with rapid polarity switching (50 ms). Nitrogen was used for curtain gas (set to 30), collision gas (set to high) and ion source gases 1 and 2 (set to 35). The source temperature was 500 °C. Spray voltage was set to −4500 V in negative mode and to 5500 V in positive mode. The values for Q1 and Q3 mass-to-charge ratios (m z−1), declustering potential, entrance potential, collision energy and collision cell exit potential were determined and optimized for each MRM for each metabolite. Ten microliters of extract were injected with PAL CTC autosampler into a 250 mm × 2.1 mm, 5-μm Luna NH2 aminopropyl HPLC column (Phenomenex) held at 25 °C for chromatographic separation. The mobile phase was solvent A: 95% water with 23.18 mM NH4OH (Sigma, Fluka Cat no. 17837-100ML), 20 mM formic acid (Sigma, Fluka Cat no. 09676-100ML) and 5% acetonitrile (pH 9.44); solvent B: 100% acetonitrile. Separation was achieved using the following gradient: 0 min—95% B, 4 min—B, 19min—2% B, 22 min—2% B, 23 min—95% B, 28 min—end. The flow rate was 300 μl min−1. All the samples were kept at 4 °C during analysis. The chromatographic peaks were identified using MultiQuant v2.1.1 (AB SCIEX), confirmed by manual inspection and the peak areas were integrated. The median of the peak area of stable isotope internal standards was calculated and used for the normalization of metabolite concentration across the samples and batches. N=6, 6.5-month-old males per group. Metabolite data were log-transformed before multivariate and univariate analyses.
Data analysis
Animals were randomized into active (suramin) and mock (saline) treatment groups at ~6 months of age. Group means and s.e.m. are reported. Behavioral data involving more than two groups were analyzed by two-way analysis of variance (ANOVA) and one-way ANOVAs (GraphPad Prism 5.0d, GraphPad Software Inc., La Jolla, CA, USA). Pair-wise post hoc testing was performed by the method of Tukey. Repeated measures ANOVA with prenatal treatment and drug as between subject factors and stimulus (mouse/cup) on time spent with mouse or cup was used as an additional test of social preference. Student’s t-test was used for comparisons involving the two groups. Significance was set at P<0.05. Bonferroni post hoc correction was used to control for multiple hypothesis testing when t-tests were used to test social preference in two or more experimental groups. Metabolomic data were analyzed using multivariate partial least squares discriminant analysis, Ward hierarchical clustering and univariate one-way ANOVA with pairwise comparisons and post hoc correction by Fisher’s least significant difference test in MetaboAnalyst.29
Results
Restoration of normal social behavior
Social behavior in mice can be quantified as the time spent interacting with a novel (‘stranger’) mouse compared with the total time spent interacting with either a mouse or a novel inanimate object.30 MIA animals showed social deficits from an early age (Figure 1a, Supplementary Figure S1a). Single-dose APT with suramin completely reversed the social abnormalities in 6.5-month-old adults (Figure 1b, Supplementary Figure S1b). Five weeks (5 half-lives) after suramin washout, a small residual benefit to social behavior was still detectable (Figure 1c, Supplementary Figure S1c). The residual social benefit of APT even after 5 weeks following suramin was correlated with retained metabolomic benefits (see below). This phenomenon was not investigated further but may be related to the development of metabolic memory and/or somatic epigenetic DNA changes that lasted longer than the physical presence of the drug.31

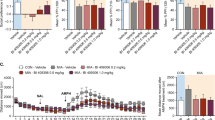
Single-dose correction of behavioral abnormalities. (a) Social abnormalities in male MIA animals were found at the earliest ages of testing at 2.25 months of age. (Student’s t-test ****P<0.0002; N=19 Saline and 25 Poly(IC)). (b) A single dose of suramin given to adult MIA mice restored normal social behavior (PIC-Sur). two-way ANOVA was first used to test for the presence of interaction between drug treatment and experimental groups. This revealed an interaction consistent with the observation that suramin benefited social behavior in the MIA animals but had no effect on normal controls (F(1,39)=13.48); P=0.0007). We then performed one-way ANOVA to test for suramin effects. A single treatment with suramin (20 mg kg−1 i.p.) given 2–4 days before testing restored normal social behavior (one-way ANOVA F(3,40)=8.95; P<0.0001; Tukey post hoc PIC-Sal versus PIC-Sur ****P<0.0001; N=8–13 per group). (c) After 5 weeks of suramin washout, the social behavior remained improved compared with saline-treated animals but was decreased from the first week after treatment. (F(3,40)=10.5; Tukey post hoc PIC-Sal versus PIC-Sur *P<0.05; N=8–13 per group). Values are expressed as means±s.e.m. (d) We estimated the strength of novelty preference32 as spontaneous alternation in the T-maze. MIA mice showed deficits in spontaneous alternation from the age of earliest testing at 4 months of age (Student’s t-test; ****P<0.0001; N=19 Saline and 25 PIC). (e) Two-way ANOVA was first used to test for the presence of interaction between drug treatment and experimental groups. This revealed an interaction consistent with the observation that suramin restored spontaneous alternation in the MIA animals but had no effect on normal controls (F(1,40)=7.609; P=0.0087). We then performed one-way ANOVA to test for suramin effects. A single dose of suramin (20 mg kg−1 i.p.) injected 2–4 days before testing corrected the deficits in young adult animals that were 5.25 months of age. (F(3,40)=9.46; ; Tukey post hoc Sal-Sal versus PIC-Sal **P<0.01; PIC-Sal versus PIC-Sur ***P<0.001); N=8–13 per group). (f) This benefit was lost after a drug washout period of 5 weeks, leaving a significant difference between control (Sal) and MIA (PIC) groups (F(3,39)=18.05; P<0.0001), but no remaining effect of suramin by post hoc testing. (Tukey post hoc PIC-Sal versus PIC-Sur P=ns; N=8–13 per group). Values are expressed as means±s.e.m. (g) Motor coordination abnormalities were quantified on the rotarod as latency to fall. Performance was abnormal from the earliest age of testing at 2.5 months of age (Student’s t-test ****P<0.0001; N=19 Saline and 25 Poly(IC)). (h) Suramin did not improve performance after two doses (20 mg kg−1 i.p.) given at 6.5 and 6.75 months of age and tested 2–4 days after the second dose. (two-way ANOVA interaction F(1,39)=0.1227; P=0.728 (ns); Poly(IC) effect F(1,39)=25.06; ****P<0.0001; treatment effect F(1,39)=0.01; P=0.908 (ns)). Values are expressed as means±s.e.m.
Restoration of spontaneous alternation in the T-maze
Novelty preference is an innate feature of normal rodent32 and human33 behavior and a predictor of socialization and communication growth in children with ASD.34 The loss or suppression of novelty preference in children with ASD is associated with the phenomenon known as insistence on sameness.35 We estimated preference for novelty as spontaneous alternation behavior in the T-maze.25 The T-maze can also be used to estimate spatial working memory, especially when food-motivated.36 We did not use the food-motivated variation in our study. We found that MIA animals showed deficient novelty preference as reflected by chance (near 50%) spontaneous alternation behavior (Figure 1d). These deficits were normalized after a single dose of suramin (Figure 1e). Five weeks after suramin washout, no residual benefit remained (Figure 1f).
Failure to restore rotarod performance in adult animals
Previous studies have shown age-dependent, postnatal loss of cerebellar Purkinje cells in the MIA model. This can reach up to 60% of Purkinje cells lost by 4 months (16 weeks) of age.23,37 Motor coordination measured by rotarod performance is deficient in the MIA model23 (and Figure 1g) and is critically dependent on the integrity of Purkinje cell circuits in the cerebellum.38 We hypothesized that since Purkinje cells are known to be lost in MIA animals by 4 months (16 weeks) of age, that APT given later in life would have no effect. Our results confirmed this. We found that a single injection of suramin given to 6-month-old adults failed to restore normal motor coordination (Figures 1g and h). Although cerebellar Purkinje cell density was not quantified in this study, our results are consistent with the notion that once Purkinje cells are lost, their function cannot be restored by APT in adult animals.
Other behaviors
Certain features of ASD and schizophrenia were not captured by our studies of the MIA model and therefore could not be interrogated for pharmacologic response to APT. For example, we did not find any abnormalities in the MIA mouse model using our protocol when we looked for certain types of anxiety-related behavior in the light–dark box paradigm (Supplementary Figure S2). Likewise, our earlier studies showed no stereotypic repetitive movements in the C57BL/6J mouse strain either by clinical observation or by testing in the hole board exploration beam break mouse behavioral pattern monitor.23 Finally, no abnormal behaviors were produced by suramin treatment itself. This was shown by the absence of behavioral differences between control mice treated with saline (‘Sal-Sal’) and those treated with suramin (‘Sal-Sur’) in Figures 1b, e and h.
Brainstem uptake of suramin
Suramin is known not to pass the blood–brain barrier;39 however, no studies have looked at suramin concentrations in areas of the brain similar to the area postrema in the brainstem that lack a blood–brain barrier.40 After completing the behavioral studies described above, we used mass spectrometry to measure drug levels in plasma, cerebrum, cerebellum and brainstem following a 5-week period of drug washout. The plasma half-life of suramin after a single dose in mice is 1 week.41 No suramin was detected in any tissue after 5 weeks of drug washout (data not shown). We next gave an acute injection of suramin (20 mg kg−1 i.p.) to the remaining subjects. After 2 days, plasma suramin was 7.64 μM±0.50, and brainstem suramin was 5.15 pmol mg−1±0.49 (Figure 2). No drug was detectable in the cerebrum or cerebellum (<0.10 pmol mg−1 wet weight) in either control (Sal-Sur) or MIA (PIC-Sur) animals, consistent with an intact blood–brain barrier that excluded suramin from these tissues. In contrast to the cerebrum and cerebellum, the brainstem showed significant suramin uptake (Figure 2). These results are consistent with the notion that nuclei in brainstem, or their projection targets in distant sites of the brain, may mediate the dramatic behavioral effects of acute and chronic APT in this model.23

Plasma and brainstem suramin quantitation. (a) Suramin is a polysulphonated napthylurea with a molecular weight of 1297 g mol−1. (b) Suramin was present in the plasma and brainstem but was not detectable in the cerebrum or cerebellum. Two days after a single 20 mg kg−1 i.p. dose of suramin, drug levels were measured in plasma (7.64 μM±0.50), brainstem (5.15 pmol mg−1±0.49), cerebrum (<0.1 pmol mg−1) and cerebellum (<0.1 pmol mg−1) in both controls (Sal) and maternal immune activation (PIC) animals. (N=4–6 per tissue). Values are expressed as means±s.e.m.
Restoration of normal purine metabolism rescues other metabolic disturbances
We analyzed the acute metabolomic effects in plasma 2 days after single-dose treatment with suramin or saline in the same animals studied behaviorally. We measured 478 metabolites from 44 pathways using mass spectrometry, analyzed the data by partial least squares discriminant analysis and visualized the results by projection in two dimensions (Figures 3a and b). This revealed sharp differences between control and MIA animals that were substantially normalized by a single treatment with suramin (Figure 3a). Figure 3b shows a similar analysis that illustrates the gradual return to disease-associated metabolism after 5 weeks of drug washout. Using hierarchical cluster analysis we found that the metabolic profiles of controls (Sal-Sal; light blue) and MIA animals that were treated with one dose of suramin (PIC-Sur; green) were more similar (major branch on the left of Figure 3c) than the metabolic profiles of saline-treated MIA animals (PIC-Sal; red) and the MIA animals tested 5 weeks after suramin washout (PIC-Sur W/O; dark blue; major branch on the right of Figure 3c). The reason that the metabolic profile had not returned completely to pretreatment conditions (to the position of the red triangles in Figure 3b) even after 5 weeks following a dose of suramin was not investigated but could be due to the development of metabolic memory and/or somatic epigenetic DNA changes that lasted longer than the physical presence of the drug.31

Metabolomic analysis. (a) APT rescues widespread metabolic abnormalities. Plasma samples were collected 2 days after a single dose of suramin (20 mg kg−1 i.p.) or saline (5 μl g−1 i.p.). This analysis shows that a single dose of suramin (PIC-Sur; green) drives the metabolism of MIA animals (PIC-Sal; red) strongly in the direction of controls (Sal-Sal; blue). Metabolomic profiles consisted of 478 metabolites from 44 biochemical pathways measured with LC-MS/MS. N=6, 6.5-month-old males per group. (b) Metabolic memory preserves metabolic rescue by APT. This analysis shows that 5 weeks after a single dose of suramin (PIC-Sur W/O; green) the metabolism of treated animals has drifted back toward that of untreated, MIA animals (PIC-Sal; red; N=6 males per group). (c) Hierarchical clustering of suramin-treated and suramin-washout metabotypes. This analysis illustrates the metabolic similarity of control (Sal-Sal; light blue) and MIA animals treated with one dose of suramin (PIC-Sur; green) compared with saline-treated MIA animals (PIC-Sal; red) and ASD-like animals tested 5 weeks after suramin washout (PIC-Sur W/O; dark blue). The numbers listed along the x axis are animal ID numbers. (d) Rank Order of metabolites disturbed in the MIA model. Multivariate analysis across the four treatment groups (PIC-Sal=MIA; PIC-Sur=acute suramin treatment; PIC-Sur w/o=5 weeks post-suramin washout; Sal-Sal=Controls). Biochemical pathway assignments are listed on the left. Relative magnitudes of each metabolite disturbance are listed on the right as high (red), intermediate (yellow or light green) and low (dark green). Variable importance in projection (VIP) scores are a multivariate statistic that reflects the impact of each metabolite on the partial least squares discriminant analysis model. VIP scores above 1.5 are significant.
Figure 3d shows the top 48 significant metabolites found in the untreated MIA animals, ranked according to their impact by variable importance in projection (VIP) score. The color-coded columns on the right of the figure indicate the direction of the change. In 43 of the 48 (90%) discriminating metabolites, suramin treatment (PIC-Sur) resulted in a metabolic shift in concentration that was either intermediate (coded yellow or light green) or in the direction of and beyond that found in control animals (Sal-Sal). The biochemical pathways represented by each metabolite are indicated on the left of Figure 3d.
Metabolic pathway analysis
The most influenced biochemical pathway in the MIA mouse was purine metabolism (Table 1). Eleven (23%) of the 48 discriminant metabolites were purines. Nine (82%) of the 11 purine metabolites were increased in the untreated MIA mice, consistent with hyperpurinergia. Only ATP and allantoin, the end product of purine metabolism in mice, were decreased in the plasma. A limitation of plasma metabolomics is that it cannot measure the effective concentration of nucleotides in the pericellular halo that defines the unstirred water layer near the cell surface where receptors and ligands meet.23 The concentration of ATP in the unstirred water layer is regulated according to conditions of cell health and danger22 in the range of 1–10 μM, which is near the EC50 of most purinergic receptors.42 This is up to 1000-fold more concentrated than the 10–20 nM levels of ATP in compartments removed from the cell surface such as the plasma.43 In the plasma we found that suramin restored 9 (82%) of the 11 purine metabolites to more normal levels, including ATP and allantoin (Figure 3d, right PIC-Sur column, coded yellow or light green) and increased inosine and deoxyinosine to above normal.
Additional pathway analysis revealed a pattern of disturbances that was remarkably similar to metabolic disturbances that have been found in children with ASDs (Table 1). Eighteen of the 44 pathways were disturbed in the MIA model. The 44 pathways interrogated by this analysis are reported in Supplementary Table S1. After purine metabolism, the next most influenced pathway was the microbiome. Microbiome metabolites are molecules that are produced by biochemical pathways that are absent in mammalian cells but are present in bacteria that reside in the gut microbiome.44 Together, purine and microbiome metabolism accounted for nearly 40% (ΣVIP=39.4%) of the impact measured by VIP scores. The two top discriminant metabolites were products of the microbiome (Figure 3d). A total of seven pathways each contributed 5% or more to the VIP pathway impact scores (Table 1). These top seven pathways were purines, microbiome metabolism, phospholipids, bile salt metabolism, sphingolipids, cholesterol, cortisol, and steroid metabolism and glycolysis. Seventy-five percent (75%) of the metabolite VIP score impact was accounted for by metabolites in these seven pathways (Table 1). Univariate statistical analysis was conducted by one-way ANOVA and pairwise group comparisons with post hoc correction (Supplementary Table S2). Forty-six (46) metabolites satisfied a false discovery rate threshold of less than 10% in this analysis. These were rank ordered by P-values. This univariate analysis identified 16 (35% of 46) metabolites (shaded yellow, Supplementary Table S3) that were also found by multivariate analysis across the four groups, and 30 (65%) additional metabolites (unshaded in Supplementary Table S2) that were discriminating only in pairwise group comparisons.
Restoration of normal purine metabolism by APT led to the concerted normalization of 17 (94%) of the 18 biochemical pathway disturbances that characterized the MIA model (Table 1; far right column). Only the bile salt pathway was not restored by suramin (Table 1,Figure 3d). The three bile salt metabolites were highest in the plasma of control animals (Figure 3d; Sal-Sal coded red in the columns on the right), lower in MIA animals (Figure 3d; PIC-Sal coded yellow) and made even lower by suramin (Figure 3d; PIC-Sur, coded dark green). Overall, we found that restoration of normal purine metabolism with APT led to the concerted improvement in both the behavioral and metabolic abnormalities in this model.
Discussion
Children with inborn errors in purine and pyrimidine metabolism have long been known to have neurodevelopmental and behavioral abnormalities.45 However, the neurochemical basis for the brain and behavioral manifestations of these classic disorders such as Lesch-Nyhan syndrome46 and adenosine deaminase deficiency45 is not yet understood. Genetic disorders of purine metabolism were some of the first single-gene disorders found to be associated with ASD behaviors. In 1969, William Nyhan described a 3-year-old boy with autistic behavior and hearing impairment caused by a defect in the first committed step in purine biosynthesis, phosphoribosyl pyrophosphate synthase.47 This mutation created an enzyme that was resistant to feedback inhibition by ATP, resulting in phosphoribosyl pyrophosphate synthase superactivity, and excess purine biosynthesis.47, 48, 49 Other reports of purine50 and pyrimidine51 disorders linked to autism soon followed.
The discovery of ATP signaling by Geoffrey Burnstock just a few years later in 1972 showed for the first time that extracellular nucleotides could act as neurotransmitters.52 However, the fields of inborn errors in purine and pyrimidine metabolism and purinergic signaling developed independently for the next 40 years. Our work in autism lies at the confluence of these two rivers of investigation. A large number of other single gene disorders,53 genetic variants, chromosomal and copy number variations2,54 and environmental factors6,55 are known to increase the risk of ASD. Most of these have no apparent ties to purine and pyrimidine metabolism. Our results are consistent with the unifying hypothesis that each factor, or mixture of factors, that causes ASD, does so by triggering a conserved cellular response to stress that we have called the CDR.22 In the MIA mouse model, the CDR is maintained by abnormalities in purinergic signaling that can be treated with antipurinergic drugs such as suramin.
It is well known that ATP inside the cell acts as an energy carrier. It is less well known that ATP and other nucleotides located outside the cell can bind to cell surface receptors and act as signaling molecules and neuromodulators that are important in inflammation,56 neurotransmission57 and many other biological processes. As such, ATP, other nucleotides and certain metabolites have been collectively considered as mitokines—signaling molecules traceable to mitochondrial function.23 Fifteen different isoforms of phosphorylated purinergic nucleotide receptors are now known.58 Four additional purinergic receptors are responsive to the unphosphorylated nucleoside adenosine, and called the P1, or adenosine receptors.59 The 15 phosphorylated nucleotide receptors are divided into seven ionotropic P2X1-7 receptors, and eight metabotropic, P2Y G-protein-coupled receptors numbered 1–14. Six former P2Y receptors (nos 3, 5 and 7–10) have been reclassified. Purinergic receptors are expressed in tissue-specific patterns on every cell type in the body. Together, the 19 P1, P2X and P2Y receptors control a broad range of biological functions that have relevance to a number of neurodevelopmental disorders, including autism, schizophrenia, attention deficit/hyperactivity and bipolar disorder. These include synaptogenesis and brain development,58 neuronal plasticity,60,61 sleep,62 regulation of the PI3K/AKT/GSK3β (glycogen synthase kinase-3β) pathway,63 control of immune responses and chronic inflammation,64 gut motility,65 gut permeability,66 taste chemosensory transduction,67 sensitivity to food allergies,68 hearing,69 innate immune signaling,70 neuroinflammation, antiviral signaling, microglial activation, neutrophil chemotaxis, autophagy, chronic pain syndromes,58 cerebellar Purkinje cell signaling and motor coordination,71 and the regulation of autonomic parasympathetic control of heart rate variability.72 The importance of metabolism in regulating behavior has recently been highlighted by the discovery that acute inhibition of the metabolic control protein GSK3β restores normal spontaneous alternation in the Y-maze in the MIA model.73
Our results show that purine metabolism is a master regulatory pathway in the MIA model (Table 1, Figure 3d, Supplementary Table S1). Correction of purine metabolism with APT restored normal social behavior (Figure 1b) and novelty preference (Figure 1e). Comprehensive metabolomic analysis revealed disturbances in several other metabolic pathways relevant to children with ASDs. These included disturbances in microbiome, phospholipid, cholesterol/sterol, sphingolipid, glycolytic and bile salt metabolism (Table 1). The top, non-microbiome-associated metabolite was quinolinic acid (Figure 3d), which was decreased in the MIA model. Quinolinic acid is a product of the indoleamine 2,3-dioxygenase pathway of tryptophan metabolism.74 Interestingly, abnormalities in purine,47,75 tryptophan,76,77 microbiome,78,79 phospholipid,80 cholesterol/sterol81 and sphingolipid82,83 metabolism have each been reported in children with ASDs. Abnormalities in purine metabolism,84 tryptophan,85 cholesterol/sterol,86 sphingolipid87 and phospholipid88 metabolism have also been described in schizophrenia. Although the detailed metabolic features of ASD and schizophrenia are different, these disorders share biochemical pathway disturbances that reveal the persistent activation of the evolutionarily conserved CDR22 in both ASD and schizophrenia. These data show that the metabolic disturbances in the MIA model and human ASD and schizophrenia are similar and provide strong support for the biochemical validity of this animal model.
Our results also reveal a novel mechanism by which drugs that cannot penetrate the blood–brain barrier can still have central nervous system effects. We show that suramin is taken up into the brainstem (Figure 2b), an area of the brain known to contain critical collections of chemosensory neurons such as the area postrema that is not protected by the blood–brain barrier.40 Purinergic neurons in the area postrema are known to integrate blood-borne signals that activate the hypothalamic–pituitary–adrenal axis during stress,89 coordinate parasympathetic and sympathetic autonomic balance90 and regulate whole-body metabolism and sickness behavior during inflammation91 in response to the evolutionarily conserved CDR associated with ASDs and other disorders.22 These data provide a plausible new mechanism that could apply to many drugs that have central nervous system effects but are known not to pass the blood–brain barrier.
Although our results of single-dose correction of abnormal behaviors in an animal model of autism and schizophrenia are encouraging, there are several caveats that must be considered before extending the results to humans. First, while the MIA mouse model captures several features of ASD and schizophrenia, no animal model can fully capture the complexities of human behavior. Second, suramin is a poor drug choice for chronic use because of potentially toxic side effects that can occur with prolonged treatment.92 Third, human forms of ASD and schizophrenia may occur by mechanisms not captured by the MIA model. Mechanisms that do not involve the CDR22 may not be amenable to APT. Human clinical trials will be necessary to answer these questions.
In summary, this study identifies purinergic signaling as a novel neurochemical switch that regulates both behavior and metabolism in the MIA mouse model. Our results provide new tools for refining current concepts of pathogenesis in autism, schizophrenia and several other neurodevelopmental disorders, and create a fresh path forward for the development of newer and safer drugs. The data provide preclinical support for the hypothesis that some of this new class of antipurinergic medicines need not be given chronically. Rather, new drugs might be given only once, or intermittently, during sensitive windows to unblock metabolism, restore more normal neural network function, improve resilience and plasticity, and permit improved development in response to behavioral and interdisciplinary therapies, and to natural play.
References
Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G et al. Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Hum Mol Genet 2012; 21: 4781–4792.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum Genet 2012; 131: 565–579.
Volk HE, Lurmann F, Penfold B, Hertz-Picciotto I, McConnell R . Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry 2013; 70: 71–77.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011; 68: 1095–1102.
Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL et al. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012; 129: e1121–e1128.
Curran LK, Newschaffer CJ, Lee LC, Crawford SO, Johnston MV, Zimmerman AW . Behaviors associated with fever in children with autism spectrum disorders. Pediatrics 2007; 120: e1386–e1392.
Vogel G . Malaria as lifesaving therapy. Science 2013; 342: 686.
Patterson PH . Infectious Behavior–Brain-Immune Connections in Autism, Schizophrenia, and Depression. The MIT Press, Cambridge, MA, USA, 2011, p162.
Mednick SA, Machon RA, Huttunen MO, Bonett D . Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry 1988; 45: 189–192.
Zerbo O, Iosif AM, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I . Is maternal influenza or fever during pregnancy associated with autism or developmental delays? Results from the CHARGE (CHildhood Autism Risks from Genetics and Environment) Study. J Autism Dev Disord 2013; 43: 25–33.
Pineda DA, Palacio LG, Puerta IC, Merchan V, Arango CP, Galvis AY et al. Environmental influences that affect attention deficit/hyperactivity disorder: study of a genetic isolate. Eur Child Adolesc Psychiatry 2007; 16: 337–346.
Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS . Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry 2013; 70: 677–685.
Sun Y, Vestergaard M, Christensen J, Nahmias AJ, Olsen J . Prenatal exposure to maternal infections and epilepsy in childhood: a population-based cohort study. Pediatrics 2008; 121: e1100–e1107.
Harvey L, Boksa P . Prenatal and postnatal animal models of immune activation: relevance to a range of neurodevelopmental disorders. Dev Neurobiol 2012; 72: 1335–1348.
Accardi MV, Daniels BA, Brown PM, Fritschy JM, Tyagarajan SK, Bowie D . Mitochondrial reactive oxygen species regulate the strength of inhibitory GABA-mediated synaptic transmission. Nat Commun 2014; 5: 3168.
Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY et al. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005; 310: 113–116.
Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH . Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 2012; 26: 607–616.
Pacheco-Lopez G, Giovanoli S, Langhans W, Meyer U . Priming of metabolic dysfunctions by prenatal immune activation in mice: relevance to schizophrenia. Schizophr Bull 2013; 39: 319–329.
Patterson PH . Modeling autistic features in animals. Pediatr Res 2011; 69: 34R–40R.
Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U . Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 2010; 35: 2462–2478.
Naviaux RK . Metabolicfeatures of the cell danger response. Mitochondrion advance online publication, 24 August 2013; PMID: 23981537.
Naviaux RK, Zolkipli Z, Wang L, Nakayama T, Naviaux JC, Le TP et al. Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS ONE 2013; 8: e57380.
Flurkey K, Currer JM, Harrison DE . Mouse models in aging research. In: Fox JG ea (ed) The Mouse in Biomedical Research, 2nd edn, vol. 3. Academic Press: San Diego, CA, USA, 2007 pp 637–672.
Frye CA, Walf AA . Effects of progesterone administration and APPswe+PSEN1Deltae9 mutation for cognitive performance of mid-aged mice. Neurobiol Learn Memory 2008; 89: 17–26.
Toth M, Gresack JE, Bangasser DA, Plona Z, Valentino RJ, Flandreau EI et al. Forebrain-specific CRF over-production during development is sufficient to induce enduring anxiety and startle abnormalities in adult mice. Neuropsychopharmacology 2014; 39: 1409–1419.
Bajad S, Shulaev V . LC-MS-based metabolomics. Methods Mol Biol 2011; 708: 213–228.
Golde WT, Gollobin P, Rodriguez LL . A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim 2005; 34: 39–43.
Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS . MetaboAnalyst 2.0–a comprehensive server for metabolomic data analysis. Nucleic Acids Res 2012; 40: W127–W133.
Moy SS, Nadler JJ, Young NB, Nonneman RJ, Segall SK, Andrade GM et al. Social approach and repetitive behavior in eleven inbred mouse strains. Behav Brain Res 2008; 191: 118–129.
Intine RV, Sarras MP Jr . Metabolic memory and chronic diabetes complications: potential role for epigenetic mechanisms. Curr Diabetes Rep 2012; 12: 551–559.
Hughes RN . Neotic preferences in laboratory rodents: issues, assessment and substrates. Neurosci Biobehav Rev 2007; 31: 441–464.
Vecera SP, Rothbart MK, Posner MI . Development of spontaneous alternation in infancy. J Cogn Neurosci 1991; 3: 351–354.
Munson J, Faja S, Meltzoff A, Abbott R, Dawson G . Neurocognitive predictors of social and communicative developmental trajectories in preschoolers with autism spectrum disorders. J Int Neuropsychol Soc 2008; 14: 956–966.
Gotham K, Bishop SL, Hus V, Huerta M, Lund S, Buja A et al. Exploring the relationship between anxiety and insistence on sameness in autism spectrum disorders. Autism res 2013; 6: 33–41.
Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP et al. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res 2007; 176: 4–20.
Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH . Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun 2009; 23: 116–123.
Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012; 488: 647–651.
Hawking F . Concentration of Bayer 205 (Germanin) in human blood and cerebrospinal fluid after treatment. Trans R Soc Trop Med Hyg 1940; 34: 37–52.
Siso S, Jeffrey M, Gonzalez L . Sensory circumventricular organs in health and disease. Acta Neuropathol 2010; 120: 689–705.
Sola F, Farao M, Pesenti E, Marsiglio A, Mongelli N, Grandi M . Antitumor activity of FCE 26644 a new growth-factor complexing molecule. Cancer Chemother Pharmacol 1995; 36: 217–222.
Jacobson KA, Balasubramanian R, Deflorian F, Gao ZG . G protein-coupled adenosine (P1) and P2Y receptors: ligand design and receptor interactions. Purinergic Signal 2012; 8: 419–436.
Adams JB, Audhya T, McDonough-Means S, Rubin RA, Quig D, Geis E et al. Nutritional and metabolic status of children with autism vs neurotypical children, and the association with autism severity. Nutr Metab 2011; 8: 34.
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 2009; 106: 3698–3703.
Micheli V, Camici M, Tozzi MG, Ipata PL, Sestini S, Bertelli M et al. Neurological disorders of purine and pyrimidine metabolism. Curr Top Med Chem 2011; 11: 923–947.
Nyhan WL . Disorders of purine and pyrimidine metabolism. Mol Genet Metab 2005; 86: 25–33.
Nyhan WL, James JA, Teberg AJ, Sweetman L, Nelson LG . A new disorder of purine metabolism with behavioral manifestations. J Pediatr 1969; 74: 20–27.
Becker MA, Raivio KO, Bakay B, Adams WB, Nyhan WL . Variant human phosphoribosylpyrophosphate synthetase altered in regulatory and catalytic functions. J Clin Invest 1980; 65: 109–120.
Nyhan WL . Phosphoribosylpyrophosphate synthetase and its abnormalities. Atlas of Inherited Metabolic Disorders. 3rd edn Hodder Arnold: London, UK, 2012, 503–506.
Ciardo F, Salerno C, Curatolo P . Neurologic aspects of adenylosuccinate lyase deficiency. J Child Neurol 2001; 16: 301–308.
Berger R, Stoker-de Vries SA, Wadman SK, Duran M, Beemer FA, de Bree PK et al. Dihydropyrimidine dehydrogenase deficiency leading to thymine-uraciluria. An inborn error of pyrimidine metabolism. Clin Chim Acta 1984; 141: 227–234.
Burnstock G, Satchell DG, Smythe A . A comparison of the excitatory and inhibitory effects of non-adrenergic, non-cholinergic nerve stimulation and exogenously applied ATP on a variety of smooth muscle preparations from different vertebrate species. Br J Pharmacol 1972; 46: 234–242.
Miles JH . Autism spectrum disorders–a genetics review. Genet Med 2011; 13: 278–294.
Betancur C . Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 2011; 1380: 42–77.
McDonald ME, Paul JF . Timing of increased autistic disorder cumulative incidence. Environ Sci Technol 2010; 44: 2112–2118.
Eltzschig HK, Sitkovsky MV, Robson SC . Purinergic signaling during inflammation. New Engl J Med 2012; 367: 2322–2333.
Burnstock G . Introduction to purinergic signalling in the brain. Adv Exp Med Biol 2013; 986: 1–12.
Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H . Purinergic signalling in the nervous system: an overview. Trends Neurosci 2009; 32: 19–29.
Chen JF, Eltzschig HK, Fredholm BB . Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov 2013; 12: 265–286.
Chen J, Tan Z, Zeng L, Zhang X, He Y, Gao W et al. Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia 2013; 61: 178–191.
Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastiao AM . Adenosine: setting the stage for plasticity. Trends Neurosci 2013; 36: 248–257.
Halassa MM . Thalamocortical dynamics of sleep: roles of purinergic neuromodulation. Semin Cell Dev Biol 2011; 22: 245–251.
Franke H, Sauer C, Rudolph C, Krugel U, Hengstler JG, Illes P . P2 receptor-mediated stimulation of the PI3-K/Akt-pathway in vivo. Glia 2009; 57: 1031–1045.
Pelegrin P . Targeting interleukin-1 signaling in chronic inflammation: focus on P2X(7) receptor and Pannexin-1. Drug News Perspect 2008; 21: 424–433.
Gallego D, Vanden Berghe P, Farre R, Tack J, Jimenez M . P2Y1 receptors mediate inhibitory neuromuscular transmission and enteric neuronal activation in small intestine. Neurogastroenterol Motil 2008; 20: 159–168.
Matos JE, Sorensen MV, Geyti CS, Robaye B, Boeynaems JM, Leipziger J . Distal colonic Na(+) absorption inhibited by luminal P2Y(2) receptors. Pflugers Arch Eur J Physiol 2007; 454: 977–987.
Surprenant A, North RA . Signaling at purinergic P2X receptors. Annu Rev Physiol 2009; 71: 333–359.
Leng Y, Yamamoto T, Kadowaki M . Alteration of cholinergic, purinergic and sensory neurotransmission in the mouse colon of food allergy model. Neurosci Lett 2008; 445: 195–198.
Housley GD, Jagger DJ, Greenwood D, Raybould NP, Salih SG, Jarlebark LE et al. Purinergic regulation of sound transduction and auditory neurotransmission. Audiol Neurootol 2002; 7: 55–61.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107.
Brockhaus J, Dressel D, Herold S, Deitmer JW . Purinergic modulation of synaptic input to Purkinje neurons in rat cerebellar brain slices. Eur J Neurosci 2004; 19: 2221–2230.
Kirchhof P, Fabritz L, Fortmuller L, Matherne GP, Lankford A, Baba HA et al. Altered sinus nodal and atrioventricular nodal function in freely moving mice overexpressing the A1 adenosine receptor. Am J Physiol Heart Circ Physiol 2003; 285: H145–H153.
Willi R, Harmeier A, Giovanoli S, Meyer U . Altered GSK3beta signaling in an infection- based mouse model of developmental neuropsychiatric disease. Neuropharmacology 2013; 73: 56–65.
Takikawa O . Biochemical and medical aspects of the indoleamine 2,3-dioxygenase- initiated L-tryptophan metabolism. Biochem Biophys Res Commun 2005; 338: 12–19.
Page T, Coleman M . Purine metabolism abnormalities in a hyperuricosuric subclass of autism. Biochim Biophys Acta 2000; 1500: 291–296.
Schain RJ, Freedman DX . Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J Pediatr 1961; 58: 315–320.
Mulder EJ, Anderson GM, Kema IP, de Bildt A, van Lang ND, den Boer JA et al. Platelet serotonin levels in pervasive developmental disorders and mental retardation: diagnostic group differences, within-group distribution, and behavioral correlates. J Am Acad Child Adolesc Psychiatry 2004; 43: 491–499.
Mulle JG, Sharp WG, Cubells JF . The gut microbiome: a new frontier in autism research. Curr Psychiatry Rep 2013; 15: 337.
Williams BL, Hornig M, Buie T, Bauman ML, Cho Paik M, Wick I et al. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS ONE 2011; 6: e24585.
Pastural E, Ritchie S, Lu Y, Jin W, Kavianpour A, Khine Su-Myat K et al. Novel plasma phospholipid biomarkers of autism: mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot Essen Fatty Acids 2009; 81: 253–264.
Tierney E, Bukelis I, Thompson RE, Ahmed K, Aneja A, Kratz L et al. Abnormalities of cholesterol metabolism in autism spectrum disorders. Am J Med Genet 2006; 141B: 666–668.
Schengrund CL, Ali-Rahmani F, Ramer JC . Cholesterol, GM1, and autism. Neurochem Res 2012; 37: 1201–1207.
Nordin V, Lekman A, Johansson M, Fredman P, Gillberg C . Gangliosides in cerebrospinal fluid in children with autism spectrum disorders. Dev Med Child Neurol 1998; 40: 587–594.
Yao JK, Dougherty GG, Reddy RD, Matson WR, Kaddurah-Daouk R, Keshavan MS . Associations between purine metabolites and monoamine neurotransmitters in first- episode psychosis. Front Cell Neurosci 2013; 7: 90.
Yao JK, Dougherty GG Jr., Reddy RD, Keshavan MS, Montrose DM, Matson WR et al. Altered interactions of tryptophan metabolites in first-episode neuroleptic-naive patients with schizophrenia. Mol Psychiatry 2010; 15: 938–953.
Bicikova M, Hill M, Ripova D, Mohr P, Hampl R . Determination of steroid metabolome as a possible tool for laboratory diagnosis of schizophrenia. J Steroid Biochem Mol Biol 2013; 133: 77–83.
Smesny S, Schmelzer CE, Hinder A, Kohler A, Schneider C, Rudzok M et al. Skin ceramide alterations in first-episode schizophrenia indicate abnormal sphingolipid metabolism. Schizophr Bull 2013; 39: 933–941.
Kaddurah-Daouk R, McEvoy J, Baillie R, Zhu H, K Yao J, Nimgaonkar VL et al. Impaired plasmalogens in patients with schizophrenia. Psychiatry Res 2012; 198: 347–352.
Lee HY, Whiteside MB, Herkenham M . Area postrema removal abolishes stimulatory effects of intravenous interleukin-1beta on hypothalamic-pituitary-adrenal axis activity and c-fos mRNA in the hypothalamic paraventricular nucleus. Brain Res Bull 1998; 46: 495–503.
Price CJ, Hoyda TD, Ferguson AV . The area postrema: a brain monitor and integrator of systemic autonomic state. Neuroscientist 2008; 14: 182–194.
McCusker RH, Kelley KW . Immune-neural connections: how the immune system's response to infectious agents influences behavior. J Exp Biol 2013; 216: 84–98.
Voogd TE, Vansterkenburg EL, Wilting J, Janssen LH . Recent research on the biological activity of suramin. Pharmacol Rev 1993; 45: 177–203.
Acknowledgements
We thank Dewleen Baker, Sophia Colamarino, Richard Haas, William Nyhan, Maya Shetreat-Klein, Leanne Chukoskie, Jeanne Townsend, Will Alaynick, Andrea Chiba, Ben Murrell, Jim Adams and Steve Edelson for helpful discussions and comments on the manuscript. We thank Laura Dugan for providing the rotarod and comments on the manuscript. We thank two anonymous reviewers for helpful comments. This research was supported by grants from the Jane Botsford Johnson Foundation (RKN) and National Institute of Health grant MH091407 (SBP), with additional support from the UCSD Christini Foundation, the Wright Family Foundation and the It Takes Guts Foundation to RKN. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Naviaux, J., Schuchbauer, M., Li, K. et al. Reversal of autism-like behaviors and metabolism in adult mice with single-dose antipurinergic therapy. Transl Psychiatry 4, e400 (2014). https://doi.org/10.1038/tp.2014.33
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.33
This article is cited by
-
Randomized clinical trial of low dose suramin intravenous infusions for treatment of autism spectrum disorder
Annals of General Psychiatry (2023)
-
Global metabolic profiles in a non-human primate model of maternal immune activation: implications for neurodevelopmental disorders
Molecular Psychiatry (2022)
-
Identification of methylation markers for diagnosis of autism spectrum disorder
Metabolic Brain Disease (2022)
-
Neurodevelopmental signatures of narcotic and neuropsychiatric risk factors in 3D human-derived forebrain organoids
Molecular Psychiatry (2021)
-
Profiles of urine and blood metabolomics in autism spectrum disorders
Metabolic Brain Disease (2021)
